ABSTRACT
We investigated transform-based algorithms for performing similarity queries in sequential databases. The transform-indexing method of Agrawal, Faloutsos, and Swami (1993) was evaluated, using both Fourier and Walsh transforms to extract features. We extended the method to enable it to handle categorical data as well as different notions of distance. Analytical and experimental evaluation of these methods was performed, using financial data and simulated genetic data.
TABLE
OF CONTENTS
4.1 Example applications and domains of data to
be mined
4.2 Numerical data versus categorical data
4.3.1 Different notions of distance
5.1 Existing sequence matching algorithms
5.2 Numerical Transform methods
III. ANALYTICAL AND EXPERIMENTAL FRAMEWORK
7. Numerical Data and Different Distance Measures
7.2 Shifting versus Shifting & Scaling
8. Numerical Data With Fourier Transform Versus Walsh Transform
9. Numerical Data Performance Results and Analysis
9.1 Frequency of false positives
10.1 Elaboration on categorical data
11. Categorical Data Results and Analysis
11.2 Find All Instances search
12. Implementation Methodology
13.1 System behavior and external control flow
V. CONCLUSIONS AND
FUTURE WORK
14.1 Transform-Indexing algorithm
14.2 Discussion of other methods
14.2.1 Dynamic Time-Warping algorithm
14.3.1 Transform-Indexing and numerical
data with Walsh transform
14.3.2 Transform-Indexing and categorical
data with Walsh transform
15.1 Dynamic Time Warping and categorical data
15.1.1 Symbol distance matrix case 1:
Boolean distances
15.1.2 Symbol distance matrix case 2:
nearly Euclidean distances
15.1.3 Symbol distance matrix case 2:
totally abstract distances
15.2 Genetic Research, the Hidden Markov Model, and
categorical data
15.3 Code: Adding algorithms and data sets to
Sequence Analyzer
A-1 Data Sets: Descriptions and Examples
I. INTRODUCTION
1. Summary
Continuing the study of the problem of finding useful patterns in large datasets, this project focuses on mining patterns from sequential data. In particular, a limited number of proposed sequence-matching algorithms were studied and compared on the basis of features such as expected speed, ease of implementation, extensibility, and correctness. This is followed by an exploration of how well some of these algorithms, which assume numerical data, are adaptable to handling certain types of non-numerical data sequences.
The "Transform-indexing" algorithm [Faloutsos, Agrawal, Swami, 1993] is implemented and tested for effectiveness in matching useful patterns in sequential data. These tests examine different definitions of “similarity” and use real data from different domains to show the performance of this method. The basic idea of the algorithm is to use a numerical transform to extract the most significant features of the data sequences, and use those extracted features as representative indexes. Faster comparisons between sequences can be done by using just the indexes to find matches, to a given degree of similarity. These results can then be pruned in a post-processing step to find the actual best match.
In the initial proposal for the Transform-indexing method, the Fourier Transform is the numerical transform method preferred. Here, the method is tested using Fourier and Walsh Transforms. The Walsh Transform is judged for effectiveness based on a comparison of results between Fourier-index tests and Walsh-index tests.
An actual implementation of the Transform and Indexing algorithm with sequences of symbols instead of real numbers tests the idea that Walsh Transforms could be used to create the indexes for specially prepared symbolic sequences. Symbolic sequences of an alphabet of size M are split into M binary sequences, which are individually transformed using the Fast Walsh Transform. Matches are defined by calculating the Euclidean distance of the indexes of these transformed sequences. The results of this distance measure are compared with the Hamming distance between sequences.
Finally, the implementation involved the creation of a GUI tool, Sequence Analyzer, which allows a user to easily test the Transform and Indexing algorithm and its experimental extensions with many different data sequences and variable algorithm parameters.
2. Role of this project
Recent advances in computing technology have enabled us to collect and record vast quantities of data, to a greater degree than seen ever before in our history. It was estimated in 1991 that the world data supply doubles every 20 months. NASA observation satellites can generate a terabyte of data per day [Fayyad and Piatetsky-Shapiro, 1996]. This unprecedented volume of data brings with it an assortment of challenges.
Awash in this sea of information, most of which will never be directly seen, researchers struggle against the growing collection of data to understand and make use of it. One of these researchers' main tools when the data under consideration is inherently sequential, including temporal data such as stock prices, is categorized as the field of sequence mining and sequence matching. It is not enough for researchers to look for exact duplication of data when comparing new observations to those previously witnessed; they must be able to quickly identify similarities in their data in order to identify data trends. In the past few years, several algorithms have been proposed to do just that. Each algorithm is attempting to achieve the most efficient and accurate means of making comparisons. The ability to make such comparisons is desirable in a wide range of data domains.
Take weather as an example. Having observed weather events and collected thorough data on weather across the globe over a span of years, the resulting amount of data is so massive that no human could possibly sort through it by looking at it directly. Yet, a forecaster wants to be able to identify the current trends in weather, classifying the current weather according to some pattern. Perhaps more intriguing is the challenge of scientists who track storms and other major weather events. If the weather data from previous years can be screened for patterns, there might be a predictive key into identifying weather phenomena which, based on such details as pressure, moisture, temperature, wind patterns, etc., could forewarn against a particular kind of major event, such as a flood, a tornado, or a tsunami.
Speaking more generally, events occur and data about them is collected. If the type of event observed is of enough interest to someone, there will be benefit in being able to compare that observed pattern with other identified patterns.
This project studies the usefulness of some of the aforementioned algorithms in comparing numerical data, which is what they were designed to do, as well as exploring how they might be applied to non-numerical data. In order to analyze these algorithms, it becomes pertinent to implement some of them. The implementation could then be studied in a variety of different scenarios , and judged by two core criteria - efficiency and accuracy. Further, variations on the algorithm can be created and tested.
This MQP provides and analyzes such an implementation. A GUI tool is also provided that can be used to test the implementation with multiple data sources and several variable algorithm settings. The GUI tool could be leveraged for further exploration of this algorithm, or to have a way to easily test the algorithm under study with different selectable data.
The foundation algorithm implementation, which relies on the use of the Discrete Fourier Transform, is then extended to alternatively allow use of the Fast Walsh Transform. The algorithm is also extended to optionally apply to similarity queries that are tolerant of affine transformations of numerical series.
An experimental extension to this algorithm, that enables the analysis of categorical (non-numerical data), is also included. This extension is based on a proposal by Professor Sergio Alvarez. The project uses this extension to match artificially generated DNA sequences, but the extension can be applied to other closed-alphabet categorical data.
The Sequence Analyzer tool created for this project is a Graphical User Interface application developed in the C++ programming language for the Windows platform and uses the Microsoft Foundation Classes. It was developed under Windows 98 in Visual Studio 6.0.
II. BACKGROUND
3. Knowledge Discovery
The amount of data available for study in many domains is so vast that it has become impossible for humans to analyze it by hand (as noted in section 2). We need a technology to be a companion to our new ability to collect and store vast amounts of data. The field of knowledge discovery in databases, commonly referred to as KDD, is concerned with acquiring knowledge from data. That is to say, the knowledge discovery process begins with raw data, and includes many steps that combine to produce an end product that is knowledge or information. The steps include data preparation, data cleaning, data mining, “incorporation of appropriate prior knowledge”, and “proper interpretation of the results of mining.” All of these steps are necessary in order for true knowledge discovery to take place; they are “essential to ensure that useful knowledge is derived from data.” [Fayyad and Piatetsky-Shapiro, 1996, p. 39]
Some of the steps incorporate intelligent ways of using computers to do the work for us that we cannot do by ourselves, in order to hasten the process, filter out noise, and so on.
3.1 Data Mining
Data mining is only a stage of the knowledge discovery process, though it is sometimes mistakenly assumed to be equivalent to KDD. Data mining done without the KDD process is often referred to as “data dredging.” Doing data mining without sufficient preparation and consideration of the data and results is not only useless, but dangerous as well, since untrue patterns and correlations can be “found”, or more correctly, falsely imposed, on data.
High level goals of the data mining step are prediction, which involves using known values of some data elements to predict unknown or future values of other data elements, and description, which means finding understandable patterns that describe the data.
These are achieved through a variety of data mining methods, which include classification (mapping a data item to a predefined class), regression (mapping data items to a resulting prediction), clustering (identifying a finite set of categories that describe the data), summarization (finding compact descriptions), dependency models (identifying dependencies between variables), and change and deviation detection (discovering significant changes compared to previously measured values). [Fayyad and Piatetsky-Shapiro, 1996, p. 44-45].
The data mining step of KDD is the step at which algorithms are actually applied to extract information from the data. Algorithms involve searching and comparing the data. The success of these algorithms requires a reasonable model representation of the data.
Depending on the complexity of the discovery needed, data mining solutions can even involve Artificial Intelligence techniques, though they do not have to.
3.2 Sequence Mining
Sequence mining is a sub-discipline of data mining which concerns data mining in sequential datasets. These are usually time series data, though not always. Sequential data can be any data for which the ordering of the elements and their position relative to each other are important. KDD coupled with sequence mining involves finding patterns, correlations, and rules that define trends, in order to make predictions or classify past behaviors.
This tends to be more complex than basic data mining because of the significance of the order of samples. In a sequential dataset, additional information beyond the mere values of the data exists inherently within the ordering of the data elements, so a question that arises is, how can that information be used most effectively in order to obtain correct and useful information?
4. Problem Domain
4.1 Example applications and domains of data to be mined
Real world applications of sequence mining are found in domains from science to investment to telecommunications. Stock market analysis is the largest use of sequence mining technology in the business domain. [Fayyad and Piatetsky-Shapiro, 1996]. A sequence mining based query of stock market data could be to find stocks that move like , for example, Microsoft, or to find a fiscal quarter in the last twenty years in which stocks were as good (or bad) for, say, agriculture as the current one.
A medical query could be to alert the doctor if there is anything dangerous or unusual in the pattern of the patient’s ECG readings. A speech recognition query could be to determine which word has just been spoken into a microphone. An image processing query could be to determine if an image is a picture of, for example, a tank or a picnic table.
An astronomical example: SETI At Home
Astronomical research is probably the application in the science domain which makes the most use of data mining. [Fayyad and Piatetsky-Shapiro, 1996]. The Search for Extra Terrestrial Intelligence Institute (SETI Institute) runs analyses on and looks for patterns in radio signals from space. One related project, supported by SETI, called SERENDIP (Search for Extraterrestrial Radio Emissions from Nearby Developed Intelligent Populations) collects data from an observatory and processes it on a 200 billion instructions per second supercomputer called SERENDIP4. The raw data is collected from the observatory at a rate of one megabyte every four minutes.
Data from the SERENDIP project is also used by the UC Berkeley SETI At Home project, which involves a client program that can be downloaded by any web user and run on a home computer. The client program takes portions of the vast store of radio signal data collected for SERENDIP. Each client program then processes this data when the computer’s resources are not being heavily used. According to the SETI Institute [SETI Institute Online], as of February 2000, SETI At Home had accumulated 1.6 million participants. Accumulated computing time contributed from May 1999 to February 2000 equaled 165,000 years. All of this data processing, which involves attempts to identify patterns in the sequential signals, uses computations of fast Fourier transforms of the original data to describe features of the data.
4.2 Numerical data versus categorical data
Numerical sequential data is sequential data which consists of information that is observed on some inherently numerical scale. Because such data contains observations that can be measured numerically, the data values can be easily represented as numbers and thus can also be manipulated and analyzed as numbers. Numerical data include features such as temperature, weight, distance, price, speed, quantity, voltage, volume, and others. Domains of such data could include stock market daily values, heat radiated by an engine, voltage observed by an electro-encephalograph, the volume of your voice, and of course many, many more, some of which are more obviously numerical than others.
This kind of data lends itself nicely to a wide range of numerical analysis options, depending on what one wishes to learn from the data. Though it is non-trivial to compare different numerical data sequences, the fact that the data is numerical makes it more straightforward to apply such analysis options than if the data were categorical. Numerical data inherently holds notions of distance between and ordering among values. It is therefore straightforward, for example, to describe the Euclidean distance between two numerical sequences. Such information is not naturally inherent to categorical data, although it may be defined explicitly in some cases.
In this context, “categorical data” is defined as data which is represented by or consists of symbols rather than numeric values. Categorical data are things like DNA sequences, consumer purchase patterns, observed sequences of commands issued by a computer user, and other information that is not inherently numerical, or cannot naturally be mapped to numerical values.
Some categorical data could be represented in different numerical ways, in order that the researcher could take advantage of the techniques available to process numerical sequences. We will explore one such representation in Section 10, as a categorical data extension to the sequence matching algorithm under study.
4.3 What is a “match”?
The “match” part of sequence-matching indicates that we are trying to identify things that are alike. The fact that we are talking about matching, as opposed to mining, means that we are not talking about discovering entirely new patterns or rules. Rather, we are taking an existing piece of data, and comparing it to a pattern which already has been established. It is implied therefore that there is going to be some specific input data sequence or sequences provided as well as some specific predefined sequences, or, templates.
4.3.1 Different notions of distance
In order to resolve whether two sequences are similar to each other or not, there must be some notion of what it means to be different. If the sequences are abstracted to be thought of as objects in an N-dimensional Euclidean space, we can define difference by saying that the further two objects are from each other in this abstract space, the less they have in common, or, the more different they are. If similarities bring the objects closer together, then two identical objects, or sequences, would occupy the same location in this space.
Thinking of similarity as proximity implies the need for a measurement of distance between objects. That is exactly how we will proceed to conceptualize difference and sameness of sequences. Similarity will be judged by measuring the distance between sequences. For example, two identical sequences would occupy the same location in our abstract space, and therefore the distance between the two would be zero.
The goal of the sequence matching algorithms we consider here is to measure the distances between sequences and find those pairs of sequences between which the distances are smallest, or are within a given margin, in order to identify positive matches or best matches between an input signal and a predefined template. The smallest distance would equate to the best match, but there are many different ways in which that distance could be measured. Sequence matching algorithms answer one of the following questions: “What pattern is this signal most like?” or “Which signals are like this pattern?”
How the distance between two sequences should be measured depends not only on the type of data being analyzed, but also on what information the observer is really looking for.
For a discussion of different types of distance, see section 8.1.
5. Current Technology
5.1 Existing sequence matching algorithms
5.1.1 Transform and Indexing
(Faloutsos, Agrawal, Swami, 1993)
This algorithm uses an indexing method to perform faster similarity queries with time series data. The method makes use of the feature extraction capability of the Discrete Fourier Transform (DFT). The DFT is used to map time series data into the frequency domain.
The algorithm makes the assumption that for most real world sequences that would be queried this way, most of the energy of the sequences are expressed in their first few frequencies, which are used as the index, and that the higher (noisier) frequencies are less relevant. They state that for much time sequence data of interest, there are a few frequencies with high amplitudes, therefore if we index only on the first few frequencies, we will have few false positives, or supposed matches that are not actually matches.
4.1.1.1 Algorithm details
The energy E(![]() ) of a sequence
) of a sequence ![]() is defined as the
sum of the energies at every point in the sequence:
is defined as the
sum of the energies at every point in the sequence:
![]()
![]()
![]()
Parseval’s Theorem allows the translation between the time domain and the frequency domain, stating that the Euclidean distance of series is preserved in such a translation. Parseval’s Theorem gives us that the energy of a sequence in the time domain is the same as its energy in the frequency domain. This relationship between domains is expressed by the following equation.
If ![]() is the Discrete
Fourier Transform of the sequence
is the Discrete
Fourier Transform of the sequence![]() , n is sequence length, t is time, and f is frequency,
Parseval’s Theorem gives us:
, n is sequence length, t is time, and f is frequency,
Parseval’s Theorem gives us:
![]()
Equation 5‑2
The numerical transform is used to extract features (strong frequencies) of sequences. These most significant features are used as indices representing the sequences. This has two major benefits. Indices are smaller to store than the original sequences, and it is faster to calculate distance between indices. It is noted that distance calculations based on a truncated list of extracted features leads to false positives. False positives are therefore removed in a post-processing step by re-running the distance test using the entire sequence rather than truncated list. The square root of the sum of squared differences is the proposed distance function between two sequences. The algorithm description specifies that the post-processing step is done in the time domain, but because of Parseval’s Theorem, this can actually be done in either the time domain or the frequency domain. It will be theoretically faster in the time domain because the results of the DFT are complex numbers, whereas the time domain values are real.
The notion of similarity in this algorithm is controlled by a tolerance parameter ε (epsilon). Two sequences are considered similar when they are within ε distance of each other. The ε parameter can be manually user defined, or determined automatically, either as a hard cutoff value, some percentage of energy, or some ratio to the average value of the sequence and/or the number of elements in the sequence.
Two categories of similarity query for sequences are defined in [Faloutsos, Agrawal, Swami, 1993]. Sub-sequence matching concerns matching parts of sequences. Whole-matching concerns only queries comparing full sequences. The Transform-Indexing paper [Faloutsos, Agrawal, Swami, 1993] only discussed whole-matching queries, though an extension to the algorithm was put forth in [Faloutsos, Ranganathan, and Manolopoulos, 1994], which handled sub-sequence queries.
Under whole-matching, two different types of query were defined. A range query takes in an input sequence and returns all sequences that are similar within ε of the input. An all pairs query finds all pairs of sequences that are within ε distance of each other.
4.1.1.3 No false dismissals
A truncation value is specified which controls the number of Fourier transform coefficients to keep as the index for a sequence. This number is typically between one and five. A partial proof is given in [Faloutsos, Agrawal, Swami, 1993] that truncating the results this way before doing the distance comparisons does not introduce any false dismissals, or matches that should occur but are instead being omitted. In sum, the proof clarifies that positive matches are defined by ε being greater than the distance value. Since more elements contributing to the distance value can only increase it, then it is impossible for a truncated distance value to be greater than ε, causing a dismissal, if the full length distance value would have been equal to or less than ε. Truncated distances will always be less than or equal to the full sequence distances.
More detail is provided on this algorithm where its implementation is discussed in Section IV.
5.1.2 Dynamic Time-Warping
(Berndt, Clifford, 1996)
4.1.2.1 Summary
Time contraction and expansion; finding shortest warping path between two series.
4.1.2.2 Problem
Though humans are good at visually detecting pattern correlations, it's hard to program machines to do the same thing - the difficulty is in matching patterns with some notion of "fuzziness."
4.1.2.3 DTW Algorithm
The algorithm proposed by Berndt and Clifford is meant for performing pattern detection on a signal, given known patterns for comparison, and uses dynamic time-warping. The dynamic "time-warping" (DTW) technique comes from the field of speech recognition. Word recognition is usually based on matching prestored word templates against an input signal of continuous speech, converted to a discrete time series. Successful recognition strategies are based on the ability to match words approximately in spite of wide variations in timing and pronunciation [Berndt and Clifford, 1996, p.231]. DTW uses a dynamic programming technique to align time series and a specific word template so that the distance between them is minimized. The time axis is stretched or compressed to achieve a reasonable fit between the template and the input signal. Because of this expansion and compression of time, the templates may match a wide variety of input signals varying in actual time lengths.
The pattern detection is done by searching a time series S for instances of a template T. A "warping path" W maps the elements of S and T such that the distance between them is minimized. That means that each element in W represents a mapping of a point in S to a point in T.
The DTW algorithm calculates and stores cumulative distances for each possible element of W. The cumulative distance of an element of W is defined as the actual distance between the two points that W is mapping plus the minimum cumulative distance from the possible previous elements in W. So, the DTW algorithm works as a best possible approximation by means of cumulative distances. Once the table of cumulative distances is filled in, the algorithm can trace through it to find the shortest possible warping path W. Refer to Section 15.1 for examples of cumulative distance calculation.
4.1.2.4 Conclusion
Berndt and Clifford observe that this algorithm may not scale well to large databases and propose that it could be used as a way of refining the results of the Transform-indexing algorithm, which is designed to perform well with large databases due to its feature extraction properties. Berndt and Clifford argue that the Transform-indexing algorithm's results are not as refined as they should be because of the likelihood of false matches inherent to the method. This argument seems to ignore the fact that the Transform-indexing algorithm includes a post-processing step designed to remove false matches, as described in sections 4.1.1 and 6 of this report.
5.1.3 Hidden Markov Model
4.1.3.1 Summary
Markov models and hidden Markov models are probabilistic predictive models of symbol generation. They are used for sequence matching by prediction; probabilities of symbols occurring in a particular sequence are based on state transitions. States generate symbols. Using a Markov Model, one can calculate the probability of any particular sequence of symbols occurring, if given a transition probability matrix.
4.1.3.2 How the Markov Model works
Markov models involve Markov Chains, which are part of the probabilistic sequence generation model. They are probabilistic finite automata, generally without input, or with input which is ignored, and with a probabilistic transition between states. Some of the states also generate one or more possible symbols.
You can graph the Markov chain, and the weights on the graph are the probabilities for transitions between states. The example in Figure 5‑1 has states q1, q2, and q3.

Figure 5‑1 Example Markov Chain
Note that the total of the weights
(probabilities) coming out of any state must equal 1.0 (100%).
The Markov assumption is that the state, or the value, at time i
depends only on the state at time i-1, and not on any historical state information
prior to that. That is to say, given a
state sequence:

P(q2|q1)=0.2 says that the probability of the next state being q2
given the current state is q1 is 0.2, or 20%. This can all be put in matrix notation. This matrix is called the Transition Matrix (T below) because it
holds information about the probabilities of transitions to and from each
state. If the total number of states is
N, T is an N ∙ N matrix.

Equation 5‑3
With its actual
values from the current example, matrix T looks like this:
 → Columns must sum to 1.0.
→ Columns must sum to 1.0.
Now say we want to know the probability of being in a particular state at
a given point in time, say t=i. First
we need to know the probability of being in that state at t = i-1. To get there, let us start at t=0.
Say for this example, the probability of starting in a given state
q is notated as Pt=0(q). Say then:
Pt=0(q1) = 0.8
Pt=0(q2) = 0.1
→ must sum to 1.0
Pt=0(q3) = 0.1
Table 5‑1 Time vector for t=0
Then, to calculate
the state probabilities for t=1, the probability (P) at t=1 of being in state
q1 is described by:
![]()
...which totals
0.4.
The calculation is also done in that fashion for Pt=1(q2) and
Pt=1(q3), which in this case are 0.29 and 0.31. Note that the sum of the probabilities of
being in any state at any given time t must equal 1.0 (such as 0.4 +
0.29 + 0.31 = 1.0).
This probability calculation can be expressed as a matrix
multiplication between the transition matrix and a time vector, in this case
for t=0, which is depicted in Table
5‑1. The dot product of the
transition matrix and the time vector for t=0 gives is the answer vector
t=1. Additionally, by multiplying the
transition matrix by the answer vector t=1, you get the answer vector for t=2,
and so on such that for any i, the answer vector for t=i is the dot product of
the transition matrix and the answer vector for t=(i-1).
Note again that the important information for determining the next
state is all contained in the current state.
All previous history is disregarded.
Hidden Markov Chains are Markov Chains with
probabilistic outputs (symbols generated), meaning several output symbols are
possible per state, each with a probability.
A Markov model is a Hidden Markov model if its states generate output
that can be seen but the sequence of the states is itself hidden, “...letting
the state of the chain generate observable data while hiding the state sequence
itself from the observer.” [Jelinek,
p.17].
4.1.3.3 Applications
In the field of sequential pattern matching, Hidden Markov
Models come into play when the question concerns either what the probability of
a specific pattern occurring is or what the probability of any one element in a
sequence being a particular element is.
This can be done as long as a state transition probability matrix is
provided.
5.2 Numerical Transform methods
5.2.1 Fourier transform
A Fourier transform is function that plots the spectrum of a signal. All wave shapes can be described exactly as combinations of sine waves of differing frequencies and varying amplitude and phase. The spectrum given by a Fourier transform represents the energy distribution of a signal as a function of frequency, specifically, of the frequencies of the component sine waves. One could also call the spectrum of a signal a set of harmonies, from harmonic analysis in music. The result of the transform is that set of harmonics. The input signal, which is graphed as a value over time, is translated into it’s frequency spectrum, which shows amplitude versus frequency. The frequency referred to here is the frequency, or repetition rate, of a particular sine wave.
In practice, an input signal may be something like this:
This signal is then separated into the different harmonics that compose it. The resulting wave forms are like the following:

and

and so on, where the frequency increases for each subsequent coefficient of the transform.
The input signal above is actually a combination of all of these different sine waves:

Each of these harmonics corresponds to one of the coefficients in the resulting array. The coefficients of a Fourier transform are complex values, not real numbers.
The calculation method used in this project for the Discrete Fourier Transform is shown in Equation 5‑5, where X is the resulting sequence, f is the frequency, x is the input signal, t is time, n is the sequence length, and j is the imaginary number (-1)0.5.
![]()
We will show a sample Fourier transform – with the following input sequence: 1854.39, 1903, 1963.95, 1966.35, 1973.66, 2000.18, 1939.08, 1948.71, 1906.12, 1981.62, 2033.71, 2033.71, 1961.76, 1964.21, 1997.16, 2062.2, 2030.36, 2104.59, 2127.19, 2130.93, 2078.69, 2119.94, 2027.66, 1988.55, 2034.21, 1935.33, 1944.18, 2044.68, 1958.6, 1954.49 The resulting Fourier coefficient magnitudes (square roots of summed squares of real and imaginary components) are: 10948.8, 195.329, 85.0428, 35.6113, 80.3047, 67.7585, 2.99938, 56.2742, 26.6221, 37.9784, 29.7078, 47.7474, 38.2915, 15.4683, 5.54336, 56.1909, then repeating symmetrically from 5.54336 ... back to 10948.8 as the last coefficient magnitude. Omitting the first Fourier coefficient and the symmetric repetition, these are graphically represented in the charts below. Note that the first coefficient is a representation of the average of the series elements (10948.8), whereas the rest of the coefficients describe the shape:
|
||
Input (value vs. time) |
Transform (amplitude vs. frequency) |


The Fourier transform can be used to extract “features” from time series data in this way. In much real world time series data which one would want to analyze, such as stock prices, the data exhibits the characteristic that most of the energy of the sequences are contained in the first few frequencies. The Fourier coefficients for those frequencies, then, can be taken as approximately representative of the entire sequence. The Discrete Fourier Transform (DFT) can be applied to numeric sequences of any length and has computational complexity on the order of O(n2) where n is the length of the sequence. A faster version of the transform, called the Fast Fourier Transform (FFT), maintains all of the same feature extraction properties but is limited to sequences whose lengths are powers of two. The Fast Fourier Transform has complexity on the order of O(nlogn), and so is preferable for long sequences where performance is a concern, provided that sequence lengths can be powers of two. For other length sequences, the FFT can be applied by padding with zeroes, but this causes some skewing of the results.
5.2.2 Walsh transform
Like the Fourier transform, the Walsh transform is a numerical process that maps time domain values into a frequency spectrum, also displaying the characteristic that the energy of many time series sequences are described by the first few coefficients of the result. Both the Walsh Transform and the Fast Walsh Transform must be applied to sequences whose lengths are powers of two. As with the FFT, sequences can be padded with zeroes to achieve a desired length, but this will skew the results of the transform.
The Walsh transform is based on the set of Walsh functions, an orthogonal set of square-wave functions that describe a Hadamard matrix. They are assumed to span a range from 0 to 1. There are only three possible values for a Walsh function: 0 outside the interval from 0 to 1, and +1 and –1 inside that interval. The number of Walsh functions used in calculating the Walsh transform is the same as the number of points in the dataset being transformed. If transforming an 8-point dataset, for example, the first 8 Walsh functions are used. These could be labeled W0(x),W1(x), W2(x), ..., W7(x) each for x = 0, ... ,1.
Different ways of defining Walsh functions are possible. For the sake of explanation, we will use the functions outlined in [Beer, 1981]. In this case, the first four Walsh functions would be graphed in the following way:
|
W0(x) |
|
|
|
|
|
W1(x) |
|
|
|
|
|
W2(x) |
|
|
|
|
|
W3(x) |
|
|
|
|
|
|
|
|
|

A compact way to represent the values of the Walsh functions is to use an N by N Hadamard matrix, where N is the number of points that will be transformed. A + represents a value of +1 and a – represents a value of –1.
The Walsh Hadamard matrix for an 8-point dataset, as defined in [Beer, 1981], is as follows. Note that sequency refers to the number of sign changes. In this representation of the Walsh functions, the sequency increases by one with each subsequent Walsh function.
W0(x)
: + + + + + + + + sequency = 0
W1(x)
: + + + + - - - - sequency = 1
W2(x) : + + - - - - + + sequency = 2
W3(x)
: + + - - + + - - sequency = 3
W4(x)
: + - - + + - - + sequency = 4
W5(x)
: + - - + - + + - sequency = 5
W6(x)
: + - + - - + - + sequency = 6
W7(x)
: + - + - + - + - sequency = 7
The sequency order for the Walsh functions represented in [Pigeon, 1998] are different:
W0(x)
: + + + + + + + + sequency = 0
W1(x)
: + + + + - - - - sequency = 1
W2(x)
: + + - - + + - - sequency = 3
W3(x)
: + + - - - - + + sequency = 2
W4(x) : + - + - + - + - sequency = 7
W5(x)
: + - + - - + - + sequency = 6
W6(x)
: + - - + + - - + sequency = 4
W7(x)
: + - - + - + + - sequency = 5
In either representation, the Walsh transform is done by applying the results of the Walsh functions to the values of the dataset being transformed. Where m is the number of points in the dataset, each of the m points is assigned a range of size 1/m between 0 and 1, and associated with the value of the Walsh function for that region. The first m Walsh functions would be used in this case, and each of the m values in the result of the transform is the sum of the products of each of the original elements with their associated Walsh function values of either +1 or –1.
To give an example, we’ll use the Hadamard matrix from [Beer]. If a is the original 8-point data sequence, element 0 of the transform A of a is the sum of each of the elements of a multiplied with +1, and:
![]()
Or much more simply, look at the Hadamard Matrix for Wj. For each +, add the corresponding element to the total. For each -, subtract the corresponding element to the total. Doing this for each j from 0 to m gives the unnormalized transform of a. We then normalize the results over the length of the sequence:
![]()
An sample transform of a 4-point dataset can be easily done by hand. Take the input series a = {1,2,3,4}. The 4x4 Hadamard matrix is:
W0
: + + + +
W1
: + + - -
W2
: + - - +
W3
: + - + -
For A0, add all the numbers together, then divide by 4:
A0
= (+1 +2 +3 +4)/4 = 10/4 = 2.5
And so on for the rest:
A1
= (+1 +2 –3 –4)/4 = -4/4 = -1
A2
= (+1 –2 –3 +4)/4 = 0/4 = 0
A3
= (+1 –2 +3 –4)/4 = -2/4 = -0.5
So the Walsh transform of {1,2,3,4} is {2.5, -1, 0, -0.5}. The values of the transform can be used to recover the original sequence by repeating the same steps but without the normalization over the length:
a1
= +2.5 +(–1) +0 +(-0.5) = 1
a2
= +2.5 +(-1) –0 –(-0.5) = 2
a3
= +2.5 -(-1) -0 +(-0.5) = 3
a4
= +2.5 -(-1) +0 –(-0.5) = 4
This recovery also works with the Walsh functions defined in [Pigeon] because in both cases the matrix is symmetric about the diagonal. The transform is calculated using the rows of the matrix, while the inverse transform is calculated using the columns of the matrix.
III. ANALYTICAL AND EXPERIMENTAL FRAMEWORK
6. Numerical Data
The numerical data used in this project includes artificially generated sequences that were created to illustrate specific characteristics of the algorithm, as well as actual daily NASDAQ market statistics spanning January 1971 to December 1999 [NASD website]. The NASDAQ statistics reflect market value of stock indexes. Market value for a single stock is calculated as the cost of a share times the number of outstanding shares. The NASDAQ indices measure stocks listed on The Nasdaq Stock Market, Inc. There is a composite index which incorporates the values of eight (to ten) different sub-indices, each of which measures stock market value of companies in its particular category. The values in the data files used in the project are the daily market values for the composite index and the sub-indices.
The primary NASDAQ index data file used in the experiments here is ALZ_NASDAQ256.alz. Each of the NASDAQ data files used in this project is named in the form ALZ_NASDAQ###.alz where “###” represents the length of the sequences contained in that file. The 256 length sequences each represent approximately one year worth of data from a NASDAQ index. The original lengths of the series vary, but are all slightly smaller than 256 elements. The sequences were each buffered with their average value to create sequences with lengths that were powers of two. In this way, we are able to use the Walsh transform to process these sequences, and therefore fairly compare the Walsh and Fourier transforms for accuracy in matching, because the same queries can be run with each type of transform.
Many of the tests described in the following sections perform different sequence matching queries with these NASDAQ index sequences. Shape templates were created to accompany the sequences of length 256. Templates are appended to the tail of the source data file.
The format of the Sequence Analyzer data files ( .alz files ) is based on comma-separated values files, with additional header information indicating sequence length and database size.
7. Numerical Data and Different Distance Measures
We define three different notions of distance measurement between sequences. Distance measurements regulate what is considered a match between two sequences. The first, called Basic, is a measurement of exact Euclidean distance. Basic is denoted by D1.
The second, Shifting distance, allows sequences with different averages to match, provided their shape is exactly the same. That is to say, this measurement allows vertical shifting. This is enabled by removing the first (index zero) coefficient of the Walsh or Fourier transform. That coefficient represents the average of the sequence, so removing enables sequences of significantly different averages to match. Shifting distance is denoted by D2.
The third, Shifting and Scaling distance, allows sequences of different averages and also different amplitudes to match, provided their essential shapes are similar. That is to say, this measurement allows vertical shifting and vertical scaling. The scaling is enabled through normalization of the test sequences. Shifting and Scaling distance is denoted by D3.
Tests were run to compare the behavior of these different distance measures and verify the correctness of their implementation . Test results are described in Sections 7.1and 7.2. One aim of this chapter is to show that these variants of distance measurement can apply effectively to the transform-indexing algorithm.
The idea of using the Transform-Indexing algorithm with these distance measure variants is supported by [Goldin and Kanellakis, 1995]. They go on to propose a syntax for specifying constraints on such variant distance measurements in time series similarity queries. They also provide a clear language for describing similarity queries and approximate matching. The following definitions and explanations are composed from information in [Goldin and Kanellakis, 1995].
A similarity transform is what is referred to in the distance definitions above as “scale” and “shift” – affine transformations of the numerical sequences, such that subtracting the shift factor and dividing by the scale factor would give the original sequence.
Where ε is the “tolerance” or “cutoff” value, an approximate match is defined as a pair of sequences that are within ε distance of each other.
A similarity match is defined as a pair of sequences that are identical after some set of similarity transforms are applied to them.
Based on these definitions, approximately similar is defined as being within ε distance after similarity transforms. Exactly similar can be defined as approximate similarity with ε set to zero.
We can say from these definitions that our implementation is concerned with approximate similarity of time series.
[Goldin and Kanellakis, 1995] states that “in many cases it is more natural to allow matching of sequences that are not close to each other in an Euclidean sense.” It is for exactly this reason that these different notions of distance are explored in this project.
The constraint syntax specified in [Goldin and Kanellakis, 1995] involves specifying an upper and lower bound for the scale and shift factors. This is generally described below:
Given a series Q, a tolerance ε, a bounds pair (la, ua) and a bounds pair (lb, ub), find all [ S, a, b ] (where S is a series) such that
D( Q, aS + b ) < ε , la < a < ua and lb < b < ub .
In our implementation, we use the unbounded cases of this query (where a and b can have any value) and do not allow such detailed user-specified constraint of scaling and shifting factors.
For approximate matching without similarity transformations (D1), we use:
Find all [ S, a, b ] such that
D( Q, aS + b ) < ε , a = 1 and b = 0 .
For shifting distance (D2), we use:
Find all [ S, b ] such that
D( Q, S + b ) < ε .
For shifting and scaling distance (D3), we use:
Find all [ S, a, b ] such that
D( Q, aS + b ) < ε .
7.1 Basic versus Shifting
Three test sequences were created to compare D1 and D2. The behavior of these two distance measurement types, by definition, is as follows.
D1 will match sequences of similar shape, as long as their averages are close enough. However, it will favor sequences whose averages are close over those whose shapes are similar. The average is represented by the first coefficient in the array that results from the Fourier transform of the original sequence. The distance value is arrived at by summing the differences between the corresponding transform coefficients of each sequence.
Given three sequences A, B, and C: if A and C are identical in shape and A and B are very different in shape, A will still match more closely to B than to C, using D1, if the averages (first coefficients) of the transform (A′) of sequence A and the transform (C′) of sequence C differ by a value that is greater than the sum of the differences between the points in A′ and B′. This is described in the equation below, where k is the number of coefficients retained in the truncated transformed sequence:
![]()
D2 will match sequences of similar shape, completely independent of what their averages are. In the example presented above, D2 would find sequences A and C to be more similar than sequences A and B.
The three test sequences are an input sequence (A) whose shape is a sine wave with a high average value, a “template” sequence (B) whose shape is a flat line with an identical average value to that of A, and a “template” sequence (C) whose shape is a sine wave with proportions identical to A but with a very different average value, much lower than that of A and B. Let D2(A,B) denote the D2 distance between sequences A and B. The defined behavior of these distance measures predicts the following result with these sequences:
D2(A,B) < D2(A,C)
AND
D3(A,B) > D3(A,C)
The original input and template sequences are shown in Figure 7‑1.

Figure 7‑1 Basic versus Shifting test input sequences
The Fourier coefficients of these sequences, including the average, are shown in Figure 8-2. Note that wherever we refer to Fourier coefficients in a graph, we actually mean the magnitudes of the coefficients. The table to the left of the figure shows the values of the coefficients, and below that, the values of the distances between sequences, where FD1(A,B) indicates Fourier-index distance between sequences named A and B.
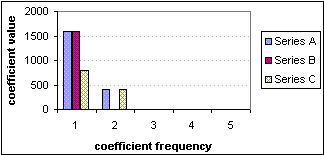
|
Fourier coefficients, BASIC |
||
|
Series A |
Series B |
Series C |
|
1600 |
1600 |
800 |
|
400 |
0 |
400 |
|
0 |
0 |
0 |
|
0 |
0 |
0 |
|
0 |
0 |
0 |
|
|
|
|
|
FD1(SeriesA,SeriesB)=25 |
||
|
FD1(SeriesA,SeriesC)=950 |
||
Figure 7‑2 Fourier coefficients - BASIC (D1)
distance
The Walsh coefficients of these sequences, including the average, are shown in Figure 8-3.

|
Walsh coefficients, BASIC |
||||
|
Series A |
Series B |
Series C |
||
|
1000 |
1000 |
50 |
||
|
31.8 |
0 |
31.8294 |
||
|
0 |
0 |
0 |
||
|
-0.391 |
0 |
-0.391 |
||
|
0 |
0 |
0 |
||
|
|
|
|
||
|
WD1(SeriesA,SeriesB)=1.98949
|
||||
|
Figure 7‑3 Walsh coefficients - BASIC (D1)
distance |
||||
The Fourier coefficients of these sequences, without the coefficient representing the average, are shown below in Figure 8-4.
Figure 7‑4 Fourier coefficients - Shifting (D2)
distance
|
|
||
|
Series A |
Series B |
Series C |
|
400 |
0 |
400 |
|
0 |
0 |
0 |
|
0 |
0 |
0 |
|
0 |
0 |
0 |
|
0 |
0 |
0 |
|
|
|
|
|
FD2(SeriesA,SeriesB)=25 |
||
|
FD1(SeriesA,SeriesC)=0.0 |
||

The Fourier coefficients of these sequences, without the coefficient representing the average, are shown below in Figure 8-5.

|
Walsh Coefficients,
SHIFTING |
||
|
Series A |
Series B |
Series C |
|
31.83 |
0 |
32.83 |
|
0 |
0 |
0 |
|
-0.391 |
0 |
-0.391 |
|
0 |
0 |
0 |
|
-0.16 |
0 |
-0.16 |
|
|
|
|
|
WD2(SeriesA,SeriesB)=1.98949
|
||
|
WD1(SeriesA,SeriesC)=0.0 |
||
Figure 7‑5 Walsh coefficients - Shifting (D2)
distance
7.2 Shifting versus Shifting & Scaling
Three test sequences were created to compare behavior of D2 and D3. In theory, D2 will match sequences of similar shape, as long as the shapes also have similar amplitudes. D3 will match shapes that are similar, regardless of their amplitude.
The test sequences include one input sequence (A), which is part of a sine wave, one “template” sequence (B) which is linear shaped but approximately the same amplitude as the input, and one “template” sequence (C) which is also part of a sine wave, but with a much smaller amplitude.
If the different distance measures behave as predicted, one would expect the following results:
D2(A,B) < D2(A,C)
AND
D3(A,B) > D3(A,C)
The input and template sequences are shown in Figure 7‑6.

Figure 7‑6 Three Similar Sequences
The magnitudes of the Fourier coefficients of these sequences (used to compute D2) are shown in Figure 7‑7.

Figure 7‑7 Fourier-index values for D2 (shift)
The normalized Fourier coefficients (used to compute D3) are shown in Figure 7‑8.

Figure 7‑8 Fourier-index values for D3 (shift & scale)
The numerical summary of the results of these tests of distance measure variants with Fourier transform indexing is shown below.
D2(A,B) = 0.139056
D2(A,C) = 0.614245
Note that D2(A,B) < D2(A,C).
D3(A,B) = 0.0183086
D3(A,C) = 0.0
Note that D3(A,B) > D3(A,C).
This fits the predicted behavior and demonstrates that these different types of distance measure are capable of finding different types of “similarity” between sequences.
The distance measurement tests for the Walsh transform require a sequence length that is a power of two. The sequences used for the Fourier test were abbreviated to a length of 32 to provide that source data for the Walsh tests. These data are shown in Figure 7‑9.

Figure 7‑9 Source data for Walsh D2 D3 tests
The shifted sequence indices of these sequences are shown in Figure 7‑10. Note that without scaling, the input sequence A, which is a partial curve, matches more closely to the straight line described in B than to the other partial curve in C. Note that although the values are negative, it is the absolute values that are used in the distance calculation, and it is the absolute values of the first few coefficients that are the furthest from zero.

Figure 7‑10 Walsh index values for D2 (shift)
As with the Fourier-index tests, the sequences here are now compared using the D3 method (scale & shift) so that sequences of similar shapes that may have different degrees of vertical scaling will match. This is again accomplished through normalization of the transform coefficients.
The normalized Walsh coefficients of the source data (used to compute D3) are shown in Figure 7‑11.

Figure 7‑11 Walsh index values for D3 (shift & scale)
In sum, the Walsh index distance test results are:
D2(A,B) = 0.03478
D2(A,C) = 0.153354
Note that D2(A,B) < D2(A,C).
D3(A,B) = 0.0364821
D3(A,C) = 0.0
Note that D3(A,B) > D3(A,C).
Note that the shifted and scaled sequence indices for Sequence A and Sequence C are identical, and thus their distances from one another is zero, even though in D2, Sequence A was closer to B than C. This is the same behavior that is observed with these distance measures using Fourier transform index values. These tests indicate two important things. First, the Transform-indexing algorithm can be adjusted to use different definitions of distance between sequence, and second, that these distance measure variants can also be used with Walsh transform indexing. Validation tests for the use of the Walsh transform as an indexing method with the Transform-indexing algorithm are described in greater detail in Section 8.
8. Numerical Data With Fourier Transform Versus Walsh Transform
Transform-indexing works by performing a numerical transformation on an input sequence, and taking the first few results, or coefficients, of the transform to be approximately representative of the whole original sequence. Whether or not a particular transform can be applied for this purpose depends on what information is actually represented by the resulting transformed values. For the Transform-indexing algorithm, the transform needs to exhibit the characteristic that most of the energy (see Equation 5‑1) of an original sequence is represented by just the first few coefficients of the transform of that sequence. Both the Fourier and Walsh transforms have this characteristic. The Fourier transform was chosen for the Transform-indexing implementation in [Faloutsos, Agrawal, Swami, 1993] and was found to be effective for that purpose. This chapter aims to use a comparison of the results of Transform-indexing with the Fourier transform and with the Walsh transform to validate the use of the Walsh transform in this capacity.
It should be noted that the coefficients of each transform are not of the same format. As distance is calculated with the transform coefficients, one would not be surprised to see some variance in the distance values produced. Walsh coefficients are real numbers, whereas Fourier coefficients are complex numbers. We may expect some minimal time efficiency improvement with the Walsh-index distance calculations over the Fourier-index calculations simply due to the fact that each element of the Fourier-index has both a real and imaginary value, and any element of the Walsh-index has only a real value.
For all of the tests in this section, the source data file was ALZ_NASDAQ256.alz. The number of series in the database is 252, which consist of 246 series that are NASDAQ category or composite values spanning most of a year, and 6 series that are for use as search templates. The search templates are zero-based row numbers 246-251. They are described below:
|
Row |
Shape
description |
|
246 |
Sine wave |
|
247 |
Line with positive slope |
|
248 |
Line with negative slope |
|
249 |
n-shaped curve – first half of a sine wave |
|
250 |
u-shaped curve – second half of a sine wave |
|
251 |
zero-valued baseline |
Table 8‑1 NASDAQ templates
The length of each series is 256. The type of search is Find All Instances (of a template). The search template used is a sine wave, located in the source file as zero-based row number 246. The results of the searches are the distances between each sequence in the database and the input (template) sequence. The first test was performed using the Fourier transform and an index size of three, with the D3 (shift & scale) distance measure. The distance results are shown in Figure 8‑1. The “unpruned”, or truncated, results are the distance values based on indices, the indices being truncated sequences of coefficients from the Fourier transform of the original data sequence. The “pruned”, or real, results are the distance values based on the full sequences of Fourier transform coefficients. Note that “real” here indicates distance calculated using a full sequence as opposed to a truncated sequence. “Real” does not imply any intrinsic or absolute distance value, but simply the distance value based on some relative definition of distance. Parseval’s theorem allows us to use the transformed sequences, instead of the originals, to compute the distances.
To give more context for the scale of the distance value results, the average distances from the zero baseline of all sequences in the ALZ_NASDAQ256.alz file (except the zero baseline itself) are given in Table 8‑2.
|
Distance measure |
Truncated Distance |
Real Distance |
|
D1 |
28.35268 |
28.44172 |
|
D2 |
1.923784 |
2.001845 |
|
D3 |
0.042349 |
0.0625 |
Table 8‑2 Average distance from zero-valued baseline

Figure 8‑1 NASDAQ – Fourier-index D3 distance from sine wave template
Figure 8‑2 shows these distances sorted by real (final) values. It may be clearer from this graph than the previous one that at no point does the truncated distance value exceed the real distance value. This example illustrates the correctness of one of the key theorems used in this algorithm – that while the indexing technique may introduce false positives, it will never cause false negatives.
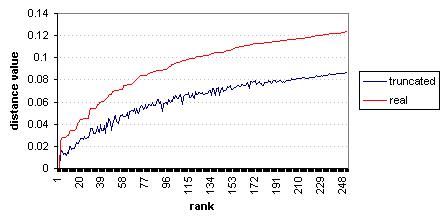
Figure 8‑2 NASDAQ – Fourier-index sorted distance values
Next we compare Walsh-indexed distances before and after pruning. As with the previous tests, the distance measure is D3 (shift & scale) and the index size is 3. What is true of truncated and real distances with the Fourier transform indexing technique also holds for the Walsh transform. This is apparent in the example in Figure 8‑3, which shows both real and truncated distances between Walsh transforms of sequences. This is a key requirement before we can safely apply the Walsh transform or any other transform to this algorithm; the indexing must not cause false dismissals. In the cases of both Fourier indexing and Walsh indexing, the actual distance values are never less than the indexed distance values because all distance measurement variants used here involve taking an absolute value of differences. In this way, the distance measurement itself is important to the prevention of false dismissals. In a sum of differences, there must never be a negative difference.

Figure 8‑3 NASDAQ – Walsh-index D3 distance values
Again, it is easier to see that index distances never exceed actual distances when looking at a graph of sorted distance values. Figure 8‑4 shows the Walsh-index distance values, sorted by the real (final) distance values.

Figure 8‑4 NASDAQ – Walsh-index sorted distance values
The distances, both truncated and real, from both Walsh and Fourier index tests, are shown in Figure 8‑5, with distances sorted by real distance values. Note that the final distances in both Walsh and Fourier tests, using the D3 (shift & scale) distance measure, are identical. This is explained by Parseval’s theorem.

Figure 8‑5 NASDAQ - Sorted Fourier and Walsh D3 distances
It can be seen in Figure 8‑5 that for the majority of sequences in the database, the Walsh-index (truncated) distance values are significantly closer to the actual (real) distance values than are the Fourier-index (truncated) distance values. The implication of this is that for post-processing performance in terms of avoiding extra calculation time due to false positives, the best choice of transform to generate indices is the Walsh transform. However, based on this test, it might be said that for lower similarity cut-off values (epsilon), the difference in performance is smaller. Note that the better (closer to 1) ranked sequences vary in which index distance, Walsh or Fourier, is closer to the actual distance. Which distance value is closer to the real distances seems to fluctuate for approximately the first 40 ranked sequences out of 250.
Whether the speed of the Walsh transform is better (faster) than the speed of the Fourier transform can not be determined conclusively from this implementation, because we implemented the n-squared complexity version of the Fourier transform (the DFT, as opposed to the FFT) and the n-log-n complexity version of the Walsh transform (FFT). The DFT was chosen over the FFT because it can apply to sequence lengths that are not powers of two. Conversely, the FWT was chosen over the n-squared complexity version of the Walsh transform because in both cases, the transform requires sequence lengths to be powers of two, so there is no reason to opt for the slower transform, as was done with the Fourier transforms. These experiments were not meant to indicate which transform, Walsh or Fourier, had better performance time based on algorithm complexity, but rather to compare how each performs relative to its role in the Transform-indexing algorithm, emphasizing such things as the characteristics of the distance values that are generated.
These experiments were also meant to illustrate whether or not the Walsh transform could be satisfactorily applied to the Transform-indexing algorithm, by illustrating that it generates query results that are at least approximately the same as those generated by using the Fourier transform. Having shown that the FWT can be applied, provided sequence lengths are powers of two, we can now use it in the experimental extension of the Transform-indexing algorithm for categorical data, described in Section 10.
9. Numerical Data Performance Results and Analysis
9.1 Frequency of false positives
The efficiency of the algorithm will vary with the choice of index size (i.e. the number of coefficients retained after truncation). Technically, the index size can be set to any number smaller than or equal to the length of the sequences. Realistically, there is no gain in having a representative index that is as long as the original sequence, and in fact, one can expect better processing time the smaller the index size is. Ideal index sizes for stock market data were shown in experiments in [Faloutsos, Agrawal, Swami, 1993] to be between 1 and 5. However, if the index size is too small, the number of false positive matches increases to a point where the post-processing time it takes to prune the results outweighs the benefit of having index-based matching in the first place. This can be resolved by experimentation and finding an optimal index size. In the experiments performed for [Faloutsos, Agrawal, Swami, 1993], they found 2 to be a satisfactory index size for most tests. However, they used only the Fourier transform to create indices. We would like to see results of similar tests with the Walsh transform indexing, as well as verifying what the optimal Fourier-index size is with our data.
Below are shown graphs of truncated and real distances with variable index sizes (1-5). Note that the area between the curves of the actual distances and the truncated distance corresponds to the likelihood of false positive matches. Figure 9‑1 shows distances for the 50 highest ranked sequences in the Find All Instances test (using the same settings as in the previous section). Distances are shown for index sizes of 1, 2, 3, 4, and 5. What is most evident about index size from this graph is that an index size of 1 gives significantly lower distance values than index sizes between 2 – 5, but that the higher index sizes are not so significantly different from each other.

Figure 9‑1 Fourier-index distances, variable index size
The Walsh-index distances for the same test are shown in Figure 9‑2.

Figure 9‑2 Walsh-index distances, variable index size
With the tolerance cutoff value (epsilon) set to 0.285, which will retrieve 2% of the database as matches to the sine wave template, we see different frequencies of false positive matches, depending on the size of the index. These results are displayed in Figure 9‑3.

Figure 9‑3 Frequency of false positive matches vs. index size
The number of false positives correlates to the time efficiency [Faloutsos, Agrawal, and Swami, 1993]. From this test, it could be said that the Fourier-index is more efficient than the Walsh when a very low index size is preferable, whereas the Walsh-index is better for index sizes of 3 or greater. The former speculation is actually inaccurate in this case only because we have implemented the Discrete Fourier Transform instead of the much more efficient Fast Fourier Transform, and therefore the Fast Walsh Transform is more efficient no matter the index size. If the Walsh and Fourier transforms that were used both had the same computational complexity (using the FFT and FWT), then the observation would hold that the Fourier-index may indeed be more time-efficient when the smallest index sizes are preferable.
9.2 Timing analysis
These results are taken from tests with the ALZ_NASDAQ256.alz database, using a Find All Instances search with the Sine Wave sequence as the search query sequence. There are 251 sequences in this database, each of length 256. The tolerance cutoff value (epsilon) used was 0.0285. Tests were run using both transform types for indexing, and using different index sizes.
An attempt was made to show a comparison between initial calculation times and post-processing time, over different index sizes and with different transform types. However, it was found that the time it takes to calculate the initial truncated distances for reasonable index sizes was too small to measure, and that therefore the initial distance calculation times, which were less than one millisecond, could not make a significant impact on an overall calculation time that varied over identical runs by as much as almost 2 seconds. It should be noted that the large variance was observed with Fourier-index runs, and that identical Walsh-index runs produced time variances of only 10 milliseconds.
A simple calculation was done to verify that the implementation of the Fourier indexing and Walsh indexing were comparatively in line with the computational complexity analyses of those transforms, and also to gather what the approximate time to perform a single computation was. With n as the sequence length, in this case 256, the complexity of the DFT should be O(n2), while the complexity of the FWT should be O(nlogn). Dividing the average time to transform input and templates with the DFT by n2 gives
45434 mSec + 65536 = 0.69326 mSec.
That divided by the number of sequences that were transformed (251) gives
0.0027620 mSec/sequence element.
To compare, dividing the average time to transform input and templates with the FWT by n gives
166 + 256 = 0.64843 mSec.
That divided by the number of sequences transformed (251) gives
0.0025834 mSec/sequence element.
The values 0.00258 and 0.00276 are close enough to one another that we can say these results agree with each other. These numbers could then be used to roughly estimate how long these operations might take for many more and much longer sequences on a comparable processor. These tests were done on a system with a Pentium 425 MHz processor and 64 megabytes of RAM.
The time in milliseconds that it took to perform the post-processing step of pruning false positives from the match results list is shown below in Figure 9‑4, with results from indexing with both the Discrete Fourier Transform (pale bars) and the Fast Walsh Transform (dark bars) over multiple index sizes.

Figure 9‑4 Time in mSec to prune false positive matches from results
These timing results are not only based on the number of false positive matches in each test (which varies between Walsh-indexing and Fourier-indexing as shown in the previous section), but also the time it takes to calculate Euclidean distance with coefficients that are complex numbers versus coefficients that are real numbers. In this particular case, these results are not especially conclusive, as the tests where the distance calculations should have taken longest were with index size 1, where the number of false positive matches was greatest, yet in both Walsh-index and Fourier-index cases, these were the tests with the smallest calculation time. Due to this seeming lack of correlation between the number of false positives and pruning distance calculation time, the observation that the Walsh-index pruning calculations seem significantly faster than the Fourier-index pruning calculations ought to be ignored. This experiment does not show conclusively what degree of timing improvement may be gained from having coefficients that are real instead of complex numbers.
10. Categorical Data
In this section we discuss
in detail what it means for data to be “categorical”, as well as how the Walsh
transform is used to extend the Transform-Indexing algorithm to apply to
categorical data series. This section
also discusses how categorical data series are generated for use with the
performance tests we run.
10.1 Elaboration on categorical data
As was explained in Section 4.2, categorical data are data which are represented by symbols instead of numbers, and includes such things as DNA sequences, consumer purchase patterns, user-issued computer system commands, and so on.
In the case of DNA sequences, which we will use as a model example, the data format we see is a series of symbols, “A”, “C”, “G”, and “T”, each of which is representative of a particular nucleotide type. The information represented in the user readable data format includes both the type of nucleotide observed, and it’s location in a sequence of nucleotides. The locations of the nucleotides in relation to one another is significant because they form tri-nucleotide sets, or amino acid codes. The amino acids specified by these codes are also in a sequence in which location relative to one another is significant, as they form together to create proteins.
So there are different ways this same data sequence could be represented. A DNA strand could be shown as a series of nucleotide symbols, or a series of symbols representing the amino acid encodings, or even a series of symbols representing kinds of proteins. We call the set of symbols used in a given dataset, or data type, the “alphabet” of that dataset or data type. It is difficult to say what the best way of representing and interpreting categorical data is, because many of the characteristics of the data points and their relationships to one another are dependent on the application domain in question. It is, therefore, non-trivial to extend the theories of sequential data matching, which traditionally assumes numeric sequences, to the problem of determining similarity of categorical sequences. In some cases, the question of similarity may be as straightforward as whether or not each symbol in a sequence is the same as the corresponding symbol in the compared sequence. That case lends itself to measuring distance with the Hamming distance of the sequences. Hamming distance is an integer measure of the number of positions in a pair of sequences where the values are not identical. In other cases, similarity may be an extremely complex and delicate issue – symbols might be weighted with different values, with a given symbol being more different from one symbol than another. In one case, the same symbol may have different weights depending on the preceding symbol. In another case, a symbol may have a particular weight dependent on the sequence of all preceding symbols, rather than just the one most previous - this last case lends itself to a state transition based model. This idea is pursued in [Kruglyak, et al, 1996]. (A description of this is included in Section 15.2.)
10.2 Walsh-index extension
We propose an extension to the Transform and Indexing algorithm which would make it applicable to some types of categorical data. The algorithm for the categorical data phase of this project relies on the use of Walsh Transforms to create representative indices for symbol sequences (of a size M alphabet). Prior to transforming, the symbol sequences are separated into M binary sequences, each of which represents whether or not one particular symbol is present at each point in the sequence, where 1 signifies the symbol is present, 0 that it isn't.
For example, a sequence of three symbols {X, Y, Z}:
Z Z Y X Y X Y Z Y X Y X Z Y X Y
Would be represented by three corresponding binary sequences, one for each symbol:
"X"
sequence:
0 0 0 1 0 1 0 0 0 1 0 1 0 0 1 0
"Y"
sequence:
0 0 1 0 1 0 1 0 1 0 1 0 0 1 0 1
"Z"
sequence:
1 1 0 0 0 0 0 1 0 0 0 0 1 0 0 0
The
original sequences are split in this way after they are read from their source
file. One character at a time, a symbol
from the original sequence is read and each time this happens a 0 or a 1 is
entered in the corresponding position of each of the symbol-specific binary
sequences.
After the sequences are split into M sequences of 0s and 1s, the binary sequences are then transformed using the Fast Walsh Transform. If K is the number of transform coefficients to keep, the first K results of the transform of each of the M sequences are calculated and stored in an index. There is one index entry for each one original sequences. The length of the index entry is K x M. These indices are used to do the quick calculation of distance between sequences, to find matches within the tolerance value epsilon distance of each other.
10.3
Data
Generation
In
order to test the Walsh-indexed categorical data extension to the Transform and
Indexing algorithm, sequences of categorical data had to be created. DNA sequence data is used as a model
sequence type, although data from actual DNA sequences is not used. Note also that the proposed means of
measuring similarity is not based on any real world similarity queries that are
made with DNA data. Further information
on actual biological applications of sequence matching technology with DNA
sequences with use of the Hidden Markov Model can be obtained from [Lander and
Green, 1987], and an extension to that in [Kruglyak, 1996].
The
point of using the DNA data type as a model is not to suggest that this method
would be directly applicable to the kinds of matching problems that molecular
biologists are trying to solve. Rather,
the point is to have a data type that is readily recognizable to most students
and researchers, providing a more grounded basis from which to discuss the
needs and possibilities of categorical sequence matching. If the data type were instead something
fabricated and completely artificial, based on a totally abstract concept of
data observations, it might limit our ability to approach the problem both
creatively and reasonably. DNA data
does have some special characteristics that make it a more interesting,
challenging, but also realistic data type to consider.
A program was written in order to generate sequences of DNA type data for observation and testing. In order that the pattern of symbols would not be completely arbitrary, the data generation tool uses the Markov Model to traverse states and generate symbols for the sequences. Generated sequences can be of any length. The lengths chosen for the generated sequences are powers of two, in order that they can be transformed using the Fast Walsh Transform. The DNA data type has a defined alphabet {A,C,G,T}. The data type also has a defined set of states, which generate these alphabet symbols, and a defined transition matrix, for specifying the probability of traversing from any one state to any state, including itself. A pseudo-random number generator is used to choose which state to traverse to from the current state, based on the transition probabilities. In this implementation, there is a start state which cannot be revisited and which produces no symbols. After exiting the start state, each state produces one of the symbols in the alphabet until the specified sequence length is reached and the program terminates. In this way, categorical sequences are generated.
The Sequence Generation code can also be adjusted to create sequences of different data types. Each such data type needs to have an alphabet, a state transition matrix, and an association between states and alphabet symbols. For example, if we chose to use amino acid sequences instead of nucleotide sequences as our DNA data representation, we could define an alphabet of 20 symbols representing each of the possible amino acids. We would then need to associate these with states and define a state transition matrix. If there are 21 states, one for each amino acid plus the start state, we need to create a 21 X 21 matrix of transition probabilities. We could then generate any length sequences of symbols from this alphabet.
These generated sequences are stored in a data file. When the file data is read by the Sequence Analyzer program, the symbol sequences are split into sequences of 0s and 1s, and indexed, as described in the previous section.
The primary source file for generated DNA sequences in this project is ALZ_DNA256_med.ALZ.
11. Categorical Data Results and Analysis
11.1 Classify Input search
The goal of this test was to verify that Classify Input search works with the categorical data extension and gives reasonable results, to provide examples of distance values between categorical series, to compare algorithm distances of sequences to Hamming distances of sequences, and to show the index/coefficient values of the individual symbol sequences [ A, C, G, T ]. The test parameters are listed below.
Source file: ALZ_DNA64_sm.alz
Sequence lengths: 64
# of template series: 10
Index size: 3
ε value: N/A
Input: Row 0
Templates: Row 1, Row 2, Row 3, Row 4, Row 5, Row 6, Row 7, Row 8, Row 9, and Row 10.
The time it took to split and transform the input sequence and ten template sequences totaled less than one millisecond. This bodes well for the efficiency of this experimental extension with small alphabet sizes. However, the similarity rank of the templates changed completely between the truncated distance calculation and the real distance calculation. That behavior essentially nullifies the usefulness of the algorithm, which aims to filter out non-matching sequences based on truncated distances. This problem is explored further in Section 11.2.
11.2 Find All Instances search
A Find All Instances search was run with a categorical data set in order to get a larger set of results to shed some light on the disappointing results of the Classify Input test. The dataset used in this test was ALZ_DNA64_lg.alz. This dataset contains 1000 sequences of length 64. The first sequence in the file was use as a comparison template for the Find All Instances query. An index size of 3 was used.
Below are the resulting truncated and real distance values from the search. Several problems immediately become apparent upon looking at the graph in Figure 11‑1. The most important of these is that there is no correlation between truncated distance values and real distance values. A scatter plot graphing the same data against one another verifies this. The similarity rank of sequences by lowest to highest distance value based on truncated coefficient indices is completely unrelated to the rank that is based on the full sequences. We have already established that the Walsh transform, which was used here, exhibits the characteristic that for non-noisy data, most of the energy of a sequence is represented by just the first few Walsh coefficients of the transform of that sequence. However, if this were the case for this dataset, we would have to see some correlation between truncated and real distances. That there is no correlation implies that the data possesses a lot of high frequency information. This makes this particular dataset unfit for this kind of representative indexing mechanism.
The second problem that is apparent on observing this figure is that the real and truncated distance values have no overlapping range; thus there is no possibility of capturing any of the real matches with a tolerance value that would filter out any of the non-matching sequences. Even the worst ranked match among the truncated distance values has a smaller distance than the best ranked real match. This problem is probably related to both the data and the indexing method. The data, being noisy and possessing lots of high frequency information will add significant weight to the distance value with higher frequency coefficients whose values were supposed to be nearly negligible and are not so. The indexing method itself may be slightly hindered in that it defines distance between sequences as the sum of the distances between each of the corresponding split sequences of the sequences being compared, for example the distance between the ‘A’ sequences plus the distance between the ‘C’ sequences, etc.. In this way, for a dataset with an alphabet size of four, the difference between the truncated distance and the real distance may be approximately quadruple what would might be normal if the data did not need to be split. This may be resolved by dividing the distance value by the alphabet size.
Of these two problems, the noisy data problem is the bigger hindrance to using the Transform-indexing algorithm for this data.

Figure 11‑1 Truncated and real distances between categorical sequences
To judge the validity of performing the distance measure that was used on this kind of data, the real Transform-Indexing distances between categorical sequences were plotted in a scatter plot versus the Hamming distances of those sequence pairs. Hamming distance is a simple integer count of the number of differences of symbols between sequences. The scatter plot is shown in Figure 11‑2. The points plot a rather straight line, indicating that there is a clear correlation between the values. This implies a direct relationship between Hamming distance and the adjusted Euclidean distance used in the Transform-indexing Categorical algorithm. So, although the distance results between these data sequences do not indicate that efficient indexing is possible or that a fixed tolerance value can be set and used for both filtering truncated sequences and capturing actual matches, we can see that the underlying distance calculation mechanism is still valid.

Figure 11‑2 Real distance vs. Hamming distance between categorical sequences
The same comparison, but using the truncated distance values, shows that there is virtually no correlation between truncated distance (with a small index size) and Hamming distance between full sequences. The vertical lines in the scatter plot in Figure 11‑3 show multiple identical distance values and are simply due to repetitions of sequences in the database.

Figure 11‑3 Truncated distance vs. Hamming distance between categorical sequences
We conclude that because it has already been established that the Walsh transform is effective at producing good representative indices from non-noisy data, it must be the case that this data contains too much high-frequency information to be effectively indexed in this way. These results do not indicate complete failure of this experimental algorithm extension, but do indicate that it is inapplicable to this data. The same experiments could be run again on other categorical data sets. It would be interesting to see if real DNA sequences possess the same degree of noise as these fabricated sequences do.
IV. SYSTEM IMPLEMENTATION
12. Implementation Methodology
12.1 Algorithm terminology
The three algorithms we have divided the original Transform-indexing algorithm into are the Fourier-index Numerical Data Transform-indexing algorithm, the Walsh-index Numerical Data Transform-indexing algorithm, and the Walsh-index Categorical Data Transform-indexing algorithm. To clarify, all of these will be referred to as “the Transform-indexing algorithm”, “the T/I algorithm”, or simply the algorithm, whenever we are speaking of common characteristics of all three.
The algorithm involves the comparison of numerical or categorical sequential data. These data sequences will be referred to as the source data, original sequences, or original series.
The comparisons of the original sequences are referred to as queries, searches, or search queries, somewhat interchangeably. In each search, there will be one original sequence which is the primary focus of the search. This sequence may be referred to as the query sequence. If the goal of the query is to find a best match for this primary sequence, based on a given set of other sequences that it is compared to, then the search type is called a Classify Input search, the query sequence is called the input sequence, and the set of other sequences are called the templates, or template sequences.
If the query sequence is instead used as a template sequence itself, and the goal is to find all instances of sequences similar to the query sequence within the database, then the search type is called a Find All Instances search. When specifying the query sequence, it is still referred to as the input sequence for this type of search. Though the goal of this search is conceptually different from Classify Input, the computations are the same, only the Find All Instances searches tend to be much more extensive and more time consuming.
Two types of numerical transform methods are possible in the implementation of the algorithm. The Discrete Fourier Transform maybe be referred to as the DFT or just Fourier transform, and the Fast Walsh Transform may be referred to as the FWT or just Walsh transform. These transforms are used to created an index to represent each sequence in a query. An index may be referred to as the truncated index, because it is a truncated list of the transform coefficients of an original sequence. Likewise it may be referred to as a coefficient index. More specifically, to differentiate between transforms, we will sometimes refer to either the Fourier-index or the Walsh-index.
Notions of distance help define whether two sequences are similar or different. A sequence is a match to the query sequence if its distance from the query sequence is within a given tolerance value (epsilon). We refer to truncated distance when we mean the distance between the truncated indices, and real distance when we mean the distance based on the full length transformed sequences or original sequences.
12.2 Major goals
There needs to be a way to run different versions of the Transform-indexing algorithm with actual data and examine the results, in order to more thoroughly study and understand the algorithm. In particular, we need to have a way to specify which version of the algorithm to run, where the data is coming from, and what kind of search query to perform.
Then we must also be able to specify our query and template sequences, and the details of the algorithm such as the tolerance value, the index size, and especially the kind of transform to used (which is related to which version of the algorithm is being run).
Next, for this to be of any use, we need a way to interpret the results, or output, of the queries. We want to see the results of the calculations, to see the rank of distance values and see which were within our tolerance, or epsilon, value, before and after post-processing. We would like to be able to visually examine the input query sequence and its matched sequences. Also, when practical, it would be interesting to examine the transform coefficients that are used to create the truncated index for each sequence.
Finally, it would be extremely convenient if tests runs could be reproduced with some parameters altered, with relatively little effort.
12.3 Solutions
A computer program called Sequence Analyzer was created to handle most of the tasks outlined above. The program is a dialog based Windows program. The first screen allows the user to specify general selections - whether to use either the numerical or categorical version of the algorithm, which type of search query to perform and where to find the source data.
In the next screen, the user is able to either launch a sample numerical query or specify algorithm variables and query and template sequences and sequence lengths. The choice of transform type (DFT or FWT) is grouped with these algorithm variables.
After a query is run, the settings are kept and from this dialog the user can adjust parameters or sequences, and then rerun the query.
The initial results of the distance calculations are displayed in a dialog after the search is performed, with indications of which sequences were preliminary matches. From this point the user can increase or decrease the tolerance value to filter out or include sequences from the post-processing step. The next screen shows a list of the actual results, that is, the sequences that were true matches and what their distances values were.
From the final dialog, the user is able to save the input and match sequences to a file in a format that can easily be copied into an Excel spreadsheet, from which graphs of the sequences can very easily be created so that the results can be examined visually. For explicit details on system behavior, see Section 13.1.
13. Sequence Analyzer Program
The next two sections focus on explaining what the required behavior of the Sequence Analyzer program is and then how this behavior is programmatically implemented. To view the C++ source files referenced in Section 13.2, please refer to the code listing in [A-3] or install the optional project directory along with the Sequence Analyzer program.
13.1 System behavior and external control flow
The user’s perspective of the program control flow and system behavior are outlined here by screen order.
Screen 1 – Main application dialog. See Figure 13‑1.
Detailed controls:
▫
Search type radio buttons - Classify Input or
Find All Instance
▫ Algorithm type drop-down list - Numerical or Categorical
▫ Database selection drop-down list - provides several choices or the option to specify another file.
Other controls:
▫ Continue button – launches Screen 2
▫ Exit button – exits application

Figure 13‑1 Screen 1
Notes:
If the option “other – specify file” is chosen under “Select Database”, a small dialog will appear when the user clicks Continue. The new file name (with its path relative to analyzer.exe) is specified here. Also note that in order to properly run an algorithm, the data file selection must be appropriate to the Algorithm type. For example, DNA files can not be used with the Numerical algorithm and NASDAQ files can not be used with the Categorical algorithm.
Screen 2 – Details dialog. See Figure 13‑2.

Figure 13‑2 Screen 2
Text fields:
Text fields in this screen provide explanations for the functions of the various Go! buttons. These are shown to the right of the associated button.
Sample control:
▫ Run Sample checkbox – selected automatically for the Numerical algorithm. This will cause a sample test query to be run after Run Search is selected. This check box is not relevant to the Categorical algorithm. When this is unselected, the Go! button for specifying algorithm parameters is enabled.
Go! controls:
These are three buttons displayed next to descriptions and all labeled “Go!”. Each one launches a new dialog in which the user specified some information needed by the program. Each button is enabled after the dialog from the previous one returns. When the last dialog returns, the Run Search button is enabled
▫ Algorithm parameters - launches algorithm details dialog for specifying algorithm variables. The algorithm details dialog has boxes in which the user specifies integer values for sequence length, number of templates, and index size, plus values for tolerance cut-off (epsilon), distance measure, and transform type. Each of these variables has a default value. If the data type is numerical, the transform type defaults to Fourier. If the data type is categorical, the transform type can only be Walsh and the distance type can only be Basic.
▫ Input sequences - launches input specification dialog. Boxes for starting row and starting column are provided, defaulting to row 0, column 0. There is also a field in which to specify a name for the input sequence for display purposes.
▫ Template sequences - launches specification dialog for templates, which are the same format as the input specification dialog. If search type is Classify Input, one dialog comes up for each template. For Find All Instances, only one template dialog comes up representing all “templates”, from which a starting row can be specified simply to cut out preceding rows from the search, and a base name can be specified, to which will be appended row numbers for each template sequence for clarity in the results display.
Once each dialog has been launched and has returned, the user can press any of the specification buttons to change algorithm parameters or sequence specifications.
Other controls:
▫ Run Search – causes algorithm to run, unless the Run Sample check box is selected, in which case a dialog comes up in which the user can make a few choices about the sample run. The next screen the user will see will be one of the several possible results screens. See descriptions of results screens for details.
▫ Quit – returns to Screen 1.
Screen 3A – Input sequences display
In some cases of the Classify Input search, a dialog will appear displaying the input and template original sequences. The user may select either OK or Cancel to continue.
Screen 3B – Transform coefficients display. See Figure 13‑3.
In a Classify Input search, a dialog will appear displaying the values of each sequence’s index. For a Fourier-index, the coefficient magnitudes are displayed. For a Walsh-index, the coefficients are displayed. For categorical data, an index whose length is the specified index size times the alphabet size is displayed. This is the concatenation of the index for each of the binary symbol sequences. For the alphabet {A,C,G,T}, the output could look like the display in Figure 13‑3 (window not shown with actual size ratio). Shown are the 1st and 2nd coefficients of the “A” series, followed by the 1st and 2nd coefficients of the “B” series, and so on. Only two coefficients are displayed for each series because that is the index size in this example. The white text boxes are not part of the display but are added here for clarification:

Figure 13‑3 Screen 3B
Screen 3C – Truncated distance results
Initial results are displayed, ordered by template row, as distances between the query sequences. An asterisk marks each distance value that is less than epsilon, the tolerance value. This does not guarantee that these marked distances will ultimately be within the tolerance range – this is only the truncated distance, used for filtering out obvious non-matches from the final processing. From here, the preliminary results can be sorted by distance value with the Sort by distance button. Also from here the tolerance value can be changed with the Change Epsilon button so that a new tolerance value is used to determine which sequences are considered in post-processing calculation and ultimately which sequences qualify as real matches.
On OK or Cancel, the program performs the post-processing step before displaying final results.
Screen 4 – Final results. See Figure 13‑4.
The final results are the names of the sequences which matched the query sequence, and their distances from the query sequence (plus Hamming distances when the data is categorical). These results are listed in whatever order they were left in by the preliminary results dialog. Note that the rank order may not be the same, so if a sorted list is desired, the user should reissue the sort command by pressing the Sort by distance button from here. From this dialog the user may do several things: sort the results, print the results and algorithm parameters to a log file with the Save results button, and print the original sequences for each of the matches and the input in columns in a data log file with the Save series button. Each of the Save buttons causes the user to be prompted for a log file name.

Figure 13‑4 Screen 4
OK returns to Screen 2 and creates any requested log file. Cancel simply returns to Screen 2.
13.2 System Architecture
The descriptions in this section assume that the reader is able to understand C code, is at least somewhat familiar with C++ programming language concepts and syntax, and but need not be very familiar with Windows programming or Microsoft Foundation Classes.
The classes in Sequence Analyzer and the purpose of each is listed in the tables below.
|
Class |
Purpose |
|
|
|
|
|
|
CAnalyzerApp |
The main application class |
|
|
CDataMatrix |
Implementation of data file I/O and internal data storage |
|
|
CFaloutsos |
Implementation class for T/I algorithm |
|
|
Complex |
Methods and arithmetic operators for Complex numbers |
|
|
timer |
Allows timestamps (in mSec) to be sent to a Timer Log file |
|
|
|
|
|
|
Dialog class |
Purpose / Dialog it defines |
Dialog resource ID |
|
|
|
|
|
CAboutDlg |
“About Sequence Analyzer” |
IDD_ABOUTBOX |
|
CAlgorithmDlg |
Algorithm parameters |
IDD_ALGORITHMDLG |
|
CAnalyzerDlg |
Screen 1 |
IDD_ANALYZER_DIALOG |
|
CDbCustomDlg |
Specify input/templates |
IDD_DBCUSTOMDLG |
|
CDetailsDlg |
Screen 2 |
IDD_DETAILSDLG |
|
CDetQueryDlg |
Specify Sample Run options |
IDD_DETQUERY |
|
CEpsilonDlg |
Select a new Epsilon value |
IDD_EPSILONDLG |
|
CFinalDlg |
Screen 4 |
IDD_FINALRESULTS |
|
CGetFileDlg |
Get “other” source file name |
IDD_GETFILE |
|
CGetLogFile |
Get results/data log name |
IDD_GETLOGNAME |
|
CShowDlg |
Screen 3A, 3B, 3C |
IDD_SHOWDLG |
Each dialog has a unique dialog resource ID by which the dialog resource is reference. If a class is defined to handle a dialog that was created in the resource editor, the class must refer to the dialog by its ID. Within the dialog class, handler functions may be implemented to capture such events as button clicks, radio button selections, and so on. A list of messages that may be handled for each dialog is available from the Class Wizard GUI.
In most cases, each class is defined in its own source file, which is named similarly as the class (dropping any additional “C” at the beginning). Thus we have, for example, Faloutsos.cpp as the source file for the CFaloutsos class and AnalyzerDlg.cpp as the source file for the CAnalyzerDlg class. Some of the related Dialog classes are grouped together in the same source file (ShowDlg.cpp).
Next we shall go through the major classes and functions in approximately the order of the control flow of the program, pointing out interesting data objects and methods along the way.
CAnalyzerApp class
CAnalyzerApp theApp – This global object representing the application itself is declared in AnalyzerApp.cpp. This object contains all of the important program variables that need to be accessed from most of the other classes. They are defined globally here so that values can be kept consistent with relative ease.
theApp global variables – The significant variables include:
int
m_nDistanceType; // Basic, Shifting, or
Shifting & Scaling
double
m_dEpsilon; // The tolerance cutoff
value
int
m_nKeepCoeff; // The index size
CString
m_csFilename; // The data source file
CString
m_csLogFileName; // File for timer log
timer
m_Timer; // A timer object to
allow time-stamping
int m_nSearchType; // Classify Input or Find All Instances
int m_nDataType; // Numeric or Categorical
int
m_nTransformType; // Fast Walsh
Transform or Discrete
//
Fourier Transform
dataspec
m_dsInput; // Starting position and
name info. for
//
the original input sequence
dataspec
* m_dsArrTemplates; // Starting
position and name info.
//
for the original template sequences
int
m_nSequenceLength; // Length of the
original sequences
int
m_nNumTemplates; // The number of
templates to compare the
//
input to.
CAnalyzerDialog class
This class defines the Screen 1 dialog which prompts the user to select a search type, an algorithm (data) type, and a source file. These values come from the various dialog controls which were created in the resource editor. Handler functions can be specified to react to user input to the controls, or the state of the controls can be ascertained when the dialog is exiting with the “Continue” button handler function. Both of these methods are used in this class in order to retrieve user options. If the “other – specify file” option is selected for the database, a CGetFileDlg dialog is launched to retrieve the file name from the user.
CDetailsDlg
This class implements the details specification dialog, from which several other dialogs are launched to acquire user input.
dataSpec – This structure is actually globally defined. This class makes use of it through the theApp object’s dataSpec variables. These hold information about the starting positions, lengths, and names of each of the original sequences that are specified by the user.
CAlgorithmDlg – This class is used for specifying algorithm parameters. An object of this class is declared by CDetailsDlg and then CAlgorithmDlg::DoModal() is called to launch the dialog. This dialog provides text fields, radio buttons, and drop down menus for the user to specify all of the algorithm remaining algorithm variables that go into theApp. The user specifies sequence length, number of templates, index size, epsilon (cutoff) value, distance measure type, and transform type. The object has public variables which receive the values of the text fields in the dialog. These fields are accessible by the calling function after DoModal() returns.
CDbCustomDlg – This class is used for specifying both input and template sequences. An object of this class is declared by CDetailsDlg and then CDbCustomDlg::DoModal() is called to launch the dialog. The text field variables are accessible in the same way as with CAlgorithmDlg. The text fields allow the user to specify a starting row, a starting column, and a name for each original sequence.
CFaloutsos myFaloutsos – This object is the main algorithm object, declared in the handler function for the “Run Search” button. Any public CFaloutsos variables that need to be set could be set after this object is declared, and before CFaloutsos::RunFaloutsos() is called.
CDataMatrix class
This class has methods for handling numerical or categorical data. The main object of the class is the data matrix itself, either m_cMatrix for a two-dimensional character array, or m_dMatrix for a two-dimensional double array.
Data is read in from the source file by _fillMatrix() or _fillCharMatrix(), each of which takes a file name as its only parameter. The class uses the header information at the top of the file to initialize the data matrix object. Then values are simply scanned in and assigned to elements of the matrix.
Single arrays of values of a specified starting row, starting column, and length, are accessed from other classes using the functions GetArray() and GetCharArray().
This class also handles data for sample runs. The class implementation is located in DataMatrix.cpp.
CFaloutsos class
RunFaloutsos() – This function’s primary purpose is simply to determine whether to run RunCategorical() or RunNumeric(). This is the entrance point into the CFaloutsos class.
RunCatagorical() – This is the main body of the algorithm when the data type is categorical. This function handles input and template series as character arrays, and transform coefficients as doubles. It also executes a splitting function which creates a set of 0/1 arrays for each alphabet symbol for each original sequence. These are the arrays that are transformed and indexed. The indices for each alphabet symbol for one original sequence are stored together in one doubles array of index size x alphabet size. Their distances between sub-indices are calculated separately and then added together.
This function uses the CDataMatrix class to retrieve data sequences, using the CDataMatrix::GetCharArray() function.
After truncated distances are calculated, the results are pruned and Hamming distances are calculated and stored in an array. All results displays are initiated from within this function.
Data
which is dynamically allocated is deallocated at the end of the function, so
the function may not return prematurely if an error is encountered.
RunNumeric() - This is the main body of the algorithm when the data type is numerical. This function handles input and template series as double arrays, and transform coefficients as doubles for the FWT and Complex values for the DFT.
This function uses the CDataMatrix class to retrieve data sequences, using the CDataMatrix::GetArray() function.
Different distance functions are provided to do distance calculations between Complex versus double values, both with and without shifting distance. If the distance measure type is Shifting & Scaling, scaling is handled by normalizing the transformed sequences before distances are calculated.
Again, displays of results are initiated from here (using _showResults() and ShowFinalResults()), and dynamically allocated data is deallocated at the end of the function.
Complex
class – The basis for this class was provided by Prof. Sergio
Alvarez. Additional methods and
operators were added as needed. The
sole use for the Complex class is to be able to represent the coefficients
of the Discrete Fourier Transform. Each
Complex object has a real and an imaginary part, which can be accessed
separately if need be. This is defined
in Complex.cpp.
Transform()
– This function takes as its one input parameter an array of doubles and returns
an array of complex values which are the DFT of the input parameter. The transform code is based on Equation 5‑5. This is called from the T_FOURIER section of
RunNumerical().
FastWalshTransform() – This function takes a source double array, and destination double array, and a length, and puts the results of the FWT of the source array into the destination array. It has no return value. This function is called from RunCategorical() and from the T_WALSH section of RunNumeric(). The code is from Steven Pigeon’s Fast Transforms module.
HammingDistance() – This function is called from RunCategorical() before final results are displayed. It takes as parameters two character arrays and a length. The return value is the number of positions where the elements of the two strings differ.
_showResults() – The _showResults() function takes an array of distances as its only parameter and, using the CFaloutsos names array member variable, formats a display with the name of the template sequence on one side and the distance value on the other. While the display string is created, tests are done on each distance value and if it is within the tolerance, an asterisk is placed next to it. Finally, a CShowDlg object is created, with its data set to the display string, and it is launched from here. _showResults() must then do a check when it returns to see if the sort button has been clicked, and if it has, set a sort flag.
ShowFinalResults() - This function prepares the Final Results dialog data and then launches that dialog. It takes two or three parameters, depending on whether the data is numerical or categorical. The first parameter as an integer representing the number of final matches. The second parameter is the full array of distances between templates sequences and the input sequence. If the data is categorical, the third parameter is the array of Hamming distance values. This function prepares the strings that will be displayed in the list box in the Final Results dialog. This is done here where we still have access to the names of the sequences. The strings are added to a list in the Final Results dialog object, and when the dialog is launched, it builds the list box with this list.
This function checks values in the dialog object when it returns to determine whether the distances need to be re-sorted and whether a data log file needs to be created with the original sequences. It sets a flag for the sorting and returns a string with the data log filename or an empty string if there is no need for a data log.
QuickSort – Quick Sort functions are defined in Faloutsos.cpp, because this is where they are needed, but they are not methods of the class. These functions are global and could therefore be used anywhere in the program if need be. They are slightly altered versions of QuickSort functions provided by Prof. Sergio Alvarez. In addition to a parameter for the array of doubles to be sorted, the QuickSort() function takes in an integer array representing the initial ordering of the array that is about to be sorted. When the doubles array elements move, their corresponding points in the ordering array also move. After sorting, the ordering array is used by other functions to reference information relative to the elements in the doubles array, such as the name associated with its value. This was done to avoid having to sort the names array and original series arrays.
Display classes and objects:
CShowDlg – This dialog class has public member variables for its title, description, and edit box content. The main purpose of the dialog is to display some kind of data. When the class object is declared, the member variables can be assigned before DoModal() is called to launch the dialog. This way the dialog class can be reused. The edit box control is the main feature of this dialog, in which the significant content is displayed. Due to the fact that it was mostly formatted numbers being displayed, this control was altered so that it always used a fixed width font. The method for doing this is not as straightforward as it seems it should be – the code to set the fixed width font comes from [Blaszcak, 1997, p. 317].
The other features of note in this dialog are the “Sort by distance” and “Change epsilon” buttons. “Change epsilon” can be pressed at any time and will bring up the CEpsilonDlg dialog, which simply prompts the user for a new numeric value for epsilon. The value is then retrieved from the CEpsilonDlg object’s variable and the global epsilon value is changed. “Sort by distance” is only relevant when distances are being displayed. This causes the dialog to return, having set a sort flag. The sort is executed by the function which initiated the CShowDlg object and a new CShowDlg dialog is displayed with the sorted values. After this button is used once it is disabled by setting a flag (m_bSortEnabled) to FALSE, which in turn causes the CWnd::ModifyStyle() method to be called.
CFinalDlg – As described under CFaloutsos::ShowFinalResults(), this dialog class builds a list of strings from a list that was initialized by the calling function. This is enabled with a public method AddToList(). Then each string is added to the CListBox control using CListBox::AddString().
Like the CShowDlg dialog, this class’s dialog has a “Sort by distance” function, which works much the same way, except that it is not disabled after one use, so when the dialog reappears with sorted values, the user may still press the button. This class also has handlers for a “Save results” button and a “Save series” button. Each of these prompts the user for a log file name. Default names are created in the handler functions based on current program variable values. The user is prompted with the GetLogFile dialog. The value of this dialog’s text field is then retrieved by CFinalDlg and returned to the calling function (in CFaloutsos) if it is a data log file. If it is a results log, the file is opened and the data displayed in the list box control is printed to the file, along with values of theApp variables.
V. CONCLUSIONS AND FUTURE WORK
14. Review
14.1 Transform-Indexing algorithm
The transform-indexing, or T/I, algorithm scales well with large databases for both memory and speed due to its feature indexing component. Its effectiveness depends on the data exhibiting the characteristic that most of its energy is concentrated in lower frequencies, less in the higher frequencies. This is essential to the indexing technique. Stock market value data exhibits this characteristic. Data that is “noisy”, or has too much energy concentrated in higher frequencies, will not be matched efficiently with this technique because the truncated indices that are created will not be representative of the whole original sequences.
The version of the algorithm that was implemented here does whole-sequence approximate matching. An extension of this algorithm exists which also does subsequence-matching. For more information on that technique, see [Faloutsos, Ranganathan, and Manolopolous, 1994].
Distance measurement that allowed scaling and shifting of sequences is more flexible than measurement that only allows shifting, or allows neither, and was found to be appropriate for the analysis of stock market data.
14.2 Discussion of other methods
We also discussed the Dynamic Time-Warping algorithm and the Hidden Markov Model. The analysis of these is summarized here.
14.2.1 Dynamic Time-Warping algorithm
This algorithm applies well to sequence matching and subsequence matching problems with numerical time-series data where the input signal is given and templates are well defined. There is no general requirement that templates and input subsequences have the same length, though that could certainly be given as a search space restriction. This algorithm can be restrictive with large databases due to memory requirements. Unlike the Transform-indexing algorithm, the Dynamic Time-warping algorithm does not rely on the data having most of its energy contained in lower frequencies. This flexibility might allow it to be applied to the kind of categorical data which we attempted to analyze with the T/I algorithm extension.
14.2.2 Hidden Markov Model
We discussed the Hidden Markov Model as a predictive pattern matching tool. The HMM uses a transition probability matrix to determine the likelihood of expressing a particular output at a particular point. This means that in order to use the HMM for sequence matching, the data would have to have an associated transition probability matrix, which restricts the domains the HMM could apply to. However, if sequences were available, a transition probability matrix could likely be constructed from these.
14.3 Extensions
14.3.1 Transform-Indexing and numerical data with Walsh transform
The Transform-indexing algorithm was successfully implemented with the Fast Walsh Transform as the sequence transforming method. The results were accurate comparative to the implementation with the Discrete Fourier Transform, and in some cases the FWT proved to be more efficient at reducing the number of false positive matches that would need to be filtered out in post-processing. The FWT performed better than the DFT with index sizes larger than two.
Also, for the worse ranked sequences in match queries, Walsh-index distances were much closer to the actual distance values than the Fourier-index distances were. This indicates that when higher tolerance values are used, the Walsh-index method will generate fewer false positive matches.
The most restrictive aspect of this transform is the requirement that sequence lengths must be powers of two (which is also the case for the Fast Fourier Transform but not the Discrete Fourier Transform). In order to analyze stock market data by year, sequences had to be buffered in order that there lengths would be powers of two and the Fourier and Walsh transform could be fairly compared for accuracy. If it is desirable to study exact sequences without buffering or truncating, and lengths of sequences are not naturally powers of two, the Walsh transform is inappropriate for indexing. Otherwise it performed very well.
14.3.2 Transform-Indexing and categorical data with Walsh transform
The Walsh transform was used to extend the Transform-indexing algorithm to apply to categorical data, using a Boolean split method. The splitting time is not prohibitive for small alphabet sizes. The transformation of multiple sequences for each original sequence was also not prohibitive for a small alphabet size because the Fast Walsh Transform is so time efficient. For extremely large alphabet sizes, this could begin to break down.
The adapted distance calculations used for this extension were shown to correlate with the Hamming distance between sequences. However, the data that was used proved unsuitable for indexing due to too much energy in the higher frequencies. Aside from the problem with the data sets, this experiment performed reasonably. It could be tried with alternate data sets.
15. Future Work
15.1 Dynamic Time Warping and categorical data
It might be possible to use the Dynamic Time-warping algorithm to extend to categorical data as was attempted with the Transform-indexing algorithm. This could be done in several different ways, depending on the particular relationship of the data symbols to each other.
This idea takes advantage of the DTW algorithm’s reliance on a cumulative distance table, and the potential to define differences between categorical symbols as numerical distance values. We will use the terminology from [] to explain cumulative distance.
The cumulative distance γ for each point represented as γ (i,j) and is defined by this recurrence relation:
γ (i,j) = δ(i,j) + min[γ(i-1,j), γ(i-1,j-1), γ(i,j-1)]
That means the cumulative distance for a point (i,j) is the actual distance between those points combined with the cumulative distance value which is the lowest among those of the three possible previous points.
A cumulative
distance table could be created for a pair of symbol sequences. Distances between symbol values could be
used, instead of Euclidean distances between points as in the T/I extension, to
calculate the distance between two categorical sequences. This could be done whether those point
distances were expressed as Boolean values, representing whether or not the
symbols were identical, or real or integer values, representing different
degrees of distance between different sequences. For the scenario with different degrees of distance, there are
two general possibilities. The first is
that the distances are based on a simple ordering of the symbols. The second is that the distances between
symbols are based on some other more abstract notion of distance which is not
readily apparent from just looking at the symbols and ordering them in a particular
way. Examples of the DTW algorithm
applied to each of these distance scenarios are given in Section 15.1.1, Section 15.1.2, and Section 15.1.3.
First, we explore a simple example from [Berndt and Clifford, 1996] which neatly illustrates how cumulative distances are calculated. Using the two mountain-shaped patterns from the [Berndt and Clifford, 1996] example, we can see the cumulative distances calculated for the points in these two series.

Figure 15‑1 Plots of two mountain-shaped patterns
As cumulative distances are calculated, they are stored in a table to avoid the need for recalculation. The cumulative distance table below [Berndt and Clifford, 1996] corresponds to the values of the series in the figure above.
|
Cumulative distance table |
||||||||
|
Mnt
10 |
6 |
90 |
50 |
70 |
110 |
130 |
90 |
70 |
|
5 |
90 |
30 |
50 |
90 |
90 |
70 |
70 |
|
|
4 |
80 |
20 |
40 |
60 |
70 |
60 |
80 |
|
|
3 |
60 |
20 |
20 |
50 |
60 |
70 |
100 |
|
|
2 |
30 |
10 |
30 |
70 |
90 |
90 |
110 |
|
|
1 |
10 |
10 |
40 |
90 |
120 |
130 |
140 |
|
|
0 |
0 |
20 |
60 |
120 |
160 |
180 |
180 |
|
|
|
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
|
Mnt 20 |
||||||||
After the table of cumulative distances is filled in, the shortest warping path can be found by tracing backwards from the last point correlation (in this case, (6,6)) and choosing the lowest value among the preceding point correlations. The shortest warping path for the series in Figure 15‑1 is given by the following sequence of correlations of (si, tj) pairs (where Mnt 20 is the S sequence and Mnt 10 is the T sequence):
W =
(6,6)(5,5)(5,4)(4,3)(3,3)(2,3)(1,2)(0,1)(0,0)
This path is indicated by bold faced values in the table above. Note that equivalent alternate decisions could have been made at points which are listed in italics.
To apply this technique to our categorical data experiment, we first define distances for the categorical symbols in each of the three test cases, and then produce cumulative distance tables for each based on the two test sequences in Section 15.1.4.
15.1.1 Symbol distance matrix case 1: Boolean distances
This is an example Boolean distance matrix – distances between symbols are only 0 or 1. A symbol is equivalent to itself and therefore the distance from itself is 0, with all other distances being 1.
|
|
u |
x |
y |
z |
|
u |
0 |
1 |
1 |
1 |
|
x |
1 |
0 |
1 |
1 |
|
y |
1 |
1 |
0 |
1 |
|
z |
1 |
1 |
1 |
0 |
15.1.2 Symbol distance matrix case 2: nearly Euclidean distances
This is an example ordering-based distance matrix – distances between symbols are based on the notion that there is a natural order to these symbols: u,x,y,z – that x comes after u and y comes after x and therefore y is further from u than x is, and so forth.
|
|
u |
x |
y |
z |
|
u |
0 |
1 |
2 |
3 |
|
x |
1 |
0 |
1 |
2 |
|
y |
2 |
1 |
0 |
1 |
|
z |
3 |
2 |
1 |
0 |
15.1.3 Symbol distance matrix case 2: totally abstract distances
This is an example abstract distance matrix – distances between symbols are not based on any readily apparent factors in the data, ordering, sameness, or otherwise.
|
|
u |
x |
y |
z |
|
u |
0.0 |
0.2 |
0.5 |
0.3 |
|
x |
0.2 |
0.0 |
0.8 |
0.4 |
|
y |
0.5 |
0.8 |
0.0 |
0.1 |
|
z |
0.3 |
0.4 |
0.1 |
0.0 |
15.1.4 Distance calculations
To illustrate how this would work, we take two randomly generated sequences, S and T, of length 6:
S = z x x y x u
T = x y y z u z
We calculate the cumulative distances between these sequences with the three different distance matrices, based on the cumulative distance definition in Equation 15‑1.
Case 1 cumulative distances:
|
T |
5 |
4 |
5 |
5 |
4 |
4 |
4 |
|
4 |
4 |
4 |
5 |
3 |
3 |
3 |
|
|
3 |
3 |
4 |
4 |
2 |
2 |
3 |
|
|
2 |
3 |
3 |
3 |
1 |
2 |
3 |
|
|
1 |
2 |
2 |
2 |
1 |
2 |
3 |
|
|
0 |
1 |
1 |
1 |
2 |
2 |
3 |
|
|
|
0 |
1 |
2 |
3 |
4 |
5 |
|
|
|
S |
||||||
W = (5,5)(4,4)(3,3)(3,2)(3,1)(2,0)(1,0)(0,0)
Case 2 cumulative distances:
|
T |
5 |
7 |
7 |
7 |
6 |
6 |
7 |
|
4 |
7 |
5 |
6 |
5 |
4 |
4 |
|
|
3 |
4 |
6 |
6 |
3 |
4 |
6 |
|
|
2 |
4 |
4 |
4 |
2 |
3 |
5 |
|
|
1 |
3 |
3 |
3 |
2 |
3 |
5 |
|
|
0 |
2 |
2 |
2 |
3 |
3 |
4 |
|
|
|
0 |
1 |
2 |
3 |
4 |
5 |
|
|
|
S |
||||||
W = (5,5)(4,4)(3,3)(3,2)(3,1)(2,0)(1,0)(0,0)
Case 3 cumulative distances:
|
T |
5 |
0.9 |
1.2 |
1.2 |
1.1 |
1.1 |
1.0 |
|
4 |
0.9 |
0.8 |
1.0 |
1.0 |
0.7 |
0.7 |
|
|
3 |
0.6 |
1.0 |
1.4 |
0.5 |
0.8 |
1.1 |
|
|
2 |
0.6 |
1.3 |
2.0 |
0.4 |
1.2 |
1.7 |
|
|
1 |
0.5 |
1.2 |
1.2 |
0.4 |
1.2 |
1.7 |
|
|
0 |
0.4 |
0.4 |
0.4 |
1.2 |
1.2 |
1.4 |
|
|
|
0 |
1 |
2 |
3 |
4 |
5 |
|
|
|
S |
||||||
W = (5,5)(4,4)(3,3)(3,2)(3,1)(2,0)(1,0)(0,0)
15.2 Genetic Research, the Hidden Markov Model, and categorical data
In
the study of human genetics, a very important area to explore is that of
multilocus linkage analysis. Different
diseases, or other genetically expressed traits, are connected to different
gene sites, or loci. One can
examine the relationship between diseases or other characteristics in this way,
through considering multiple loci simultaneously.
Linkage
is the implication that two (or more) things, such as disease
conditions, are related in that they are coded for on the same chromosome, and
that therefore the presence of one disease condition may be a warning flag for
the possible presence of the other. Also,
a higher frequency of encountering two traits or diseases being expressed in
the same individual may suggest a nearer location of the sites of the encodings
for those disease conditions on the shared chromosome. Likewise, traits that are less frequently
encountered together may be have chromosome encodings located further apart.
A major problem with performing linkage
analysis is the lack of complete information. Important data are often missing
or imperfect. An additional
computational obstacle is that in doing a multilocus analysis, computing time
scales exponentially based on the number of loci being analyzed.
Hidden Markov Models have been used in
genetic reconstruction algorithms.
Hidden Markov Models can be used to predict patterns or outputs based on
a transition probability matrix. In the
genetic reconstruction algorithm, the “inheritance distribution” is considered
to be a Hidden Markov process. See
[Lander and Green, 1987] and [Kruglyak, 1996].
This
existing algorithm could be explored further from the perspective of
implementations of sequence mining algorithms, as a good real world example of
how Hidden Markov Models can be used for pattern matching. From that basis one might extend the use of the
Hidden Markov Model to pattern matching
in other domains, specifically other sequential categorical data domains.
15.3 Code: Adding algorithms and data sets to Sequence Analyzer
In the Sequence Analyzer program, the sequence data is separate from the algorithm code. The Transform-indexing algorithm is almost entirely encompassed in the CFaloutsos class. External to that class, the only T/I-specific code is the calls to CFaloutsos methods to initialize the class object’s data. This is done from the CDetailsDlg class, which could quite easily be changed to initialize and launch different algorithms based on a specification from a drop-down menu in the first application dialog.
In order to add another algorithm, the algorithm would need to be implemented as its own class. Its initialization could happen parallel to the way CFaloutsos initialization happens, with sequence definitions specified outside of the start of the algorithm. A CNewAlgorithm::RunNewAlgorithm() method could be called for it from CDetailsDlg::OnRunSearchClicked() in the same way that CFaloutsos::RunFaloutsos() is called currently.
To change the list of data sets in the program, string labels for new data sets must be added to the data set Combo Box control in CAnalyzerDlg. In the “Continue” button handler, these labels must be associated with the files they represent. If an absolute path is not given, the data file must exist within the same directory as the program executable file. Alternately, a different data set file may be specified by the program user by selecting the option “Other – specify file”, provided the file specified is of the correct format.
A standard numerical data file for Sequence Analyzer is a text file with series in rows, with elements separated by commas without spaces or tabs, and with equal numbers of elements per row. The number of rows and the length of sequences is specified at the head of the file.
For categorical data, the data set’s alphabet is also specified at the head of the file, and the sequence elements are contiguous characters that are not separated by commas, spaces, tabs, or any other delimiter. Series are still in rows, separated by line breaks.
In order to change the format definition of the data files, the way files are parsed in the CDataMatrix class must be altered. The format of the file does not affect any other part of the program, as long as the data is the same and the CDataMatrix class holds the values for the number of rows, the length of sequences, and the alphabet definition where appropriate.
VI. BIBLIOGRAPHY
Agrawal, Rakesh, Faloutsos, Christos, and Swami, Arun, "Efficient Similarity Search In Sequence Databases," Foundations of Data Organization and Algorithms (FODO) Conference, 1993.
Beer, Tom, "Walsh Transforms," American Journal of Physics, vol 49 no 5, May 1981, pp466-472.
Berndt, Donald J. and Clifford, James, "Finding Patterns in Time Series: A Dynamic Programming Approach," Advances in Knowledge Discovery and Data Mining, pp229-248, AAAI Press, 1996.
Blaszcak, Mike, Professional MFC with Visual C++ 5.0, Wrox Press Ltd, 1997.
Faloutsos, Christos, Ranganathan, M., and Manolopoulos, Yannis, "Fast Subsequence Matching in Time Series Databases," Proceedings of the ACM (SIGMOD),1994.
Fayyad, Usama, Piatetsky-Shapiro, Gregory, and Smyth, Padhraic, "From Data Mining to Knowledge Discovery in Databases," AI Magazine, Fall 1996, pp37-54.
Goldin, Dina Q. and Kanellakis, Paris C., “On similarity queries for time series data: Constraint specification and implementation,” Proceedings of Constraint Programming 95, Marseilles, France, 1995.
Jelinek, Frederick, Statistical Methods for Speech Recognition, MIT Press 1997, pp15-37.
Kruglyak, Leonid, et al, "Parametric and Nonparametric Linkage Analysis: A Unified Multipoint Approach," American Journal of Human Genetics, vol 58, 1996, pp1347-1363.
Lander, E.S., and Green, P., "Construction of Multilocus Genetic Maps in Humans," Proc Natl Acad Sci USA vol 84 1987 pp2363-2367.
National
Association of Securities Dealers, Inc
website: http://www.nasd.com/ ,
© Copyright 2000
Pigeon, Stephen, “Le Transformées Rapides (Fast Transforms)”, 1998, http://www.iro.umontreal.ca/~pigeon/Wavelets/TransformeesRapides.html
Rabiner, Lawrence R., "A Tutorial on Hidden Markov Models and Selected Applications in Speech recognition," Proceedings of the IEEE, vol. 77, no. 2, February 1989 pp257-286.
SETI Institute Online, SETI Institute, http://www.seti.org/
Srikant, Ramakrishnan, and Agrawal, Rakesh, "Mining Sequential Patterns: Generalizations and Performance Improvements," Proceedings of the 5th International Conference on Extending Database Technology (EDBT), Avignon, France, 1996.
Yi, Byoung-Kee, Jagadish, H. V., and Faloutsos, Christos, “Efficient Retrieval of Similar Time Sequences Under Time Warping,” International Conference of Data Engineering, 1998.